
Significance
The oxidation of dissolved Fe(II) to Fe(III) plays an important role in shaping the chemistry of natural waters. This process drives the formation of poorly crystalline Fe(III) precipitates that act as reactive surfaces and colloidal carriers for various solutes, including phosphate (PO₄³⁻), organic carbon (OC), and trace metals. These precipitates determine how nutrients and contaminants are immobilized or mobilized, influencing eutrophication dynamics and carbon sequestration. Yet, in near-neutral environments, the structure and reactivity of these Fe(III) solids are rarely simple; they evolve in response to the chemical interplay among phosphate, silicate, calcium, and dissolved organic matter (DOM). Each of these constituents modulates Fe(III) polymerization, particle aggregation, and the stability of colloidal species in unique but interdependent ways. Although phosphate’s affinity for Fe(III) is well recognized—it readily co-precipitates as amorphous Fe(III)–phosphate or Ca–Fe(III)–phosphate—the presence of organic ligands complicates these pathways. Organic acids, humic substances, and other natural ligands can either inhibit Fe(III) polymerization or redirect it toward mixed Fe–organic phases with altered sorptive behavior. Previous studies have shown that such ligands reduce crystallinity, generate Fe(III)–organic complexes, and, under some conditions, even enhance phosphate retention through surface modification. However, the outcome depends sensitively on ligand identity, its Fe(III)-binding strength, and its concentration relative to Fe. This chemical complexity has left large gaps in predicting how Fe(III) precipitates control phosphorus and organic carbon cycling under realistic environmental conditions. Earlier research established that phosphate and calcium together yield mixed Ca–Fe(III)–phosphates with higher Fe polymerization, while phosphate scarcity favors ferrihydrite and lepidocrocite formation. Similarly, natural organic matter can stabilize amorphous Fe phases, but its effect is contingent upon molecular composition. Despite these insights, few controlled experiments have addressed the combined impact of multiple ligands—especially those differing in molecular weight and coordination chemistry—under the buffered, near-neutral conditions typical of natural waters.
To this account, new research paper published in Environmental science. Processes & impacts and conducted by Dr. Ville Nenonen, Dr. Ralf Kaegi, Dr. Stephan Hug, Dr. Jörg Luster, Dr. Jörg Göttlicher, Dr. Stefan Mangold, and led by Professor Lenny Winkel and Professor Andreas Voegelin from the Eawag, Swiss Federal Institute of Aquatic Science and Technology, the researchers developed two conceptual models: one describing Fe(III) precipitation in the presence of weak organic ligands that favor crystalline Fe–phosphate phases, and another illustrating how strong organic ligands and calcium induce amorphous, colloidal Fe–organic–phosphate aggregates. These models integrate ligand binding strength, OC/Fe ratio, and PO₄/Fe ratio as predictive parameters for Fe(III)-precipitate structure. Together, they offer a mechanistic framework that connects nanoscale mineral transformations to phosphorus and carbon cycling in natural waters.
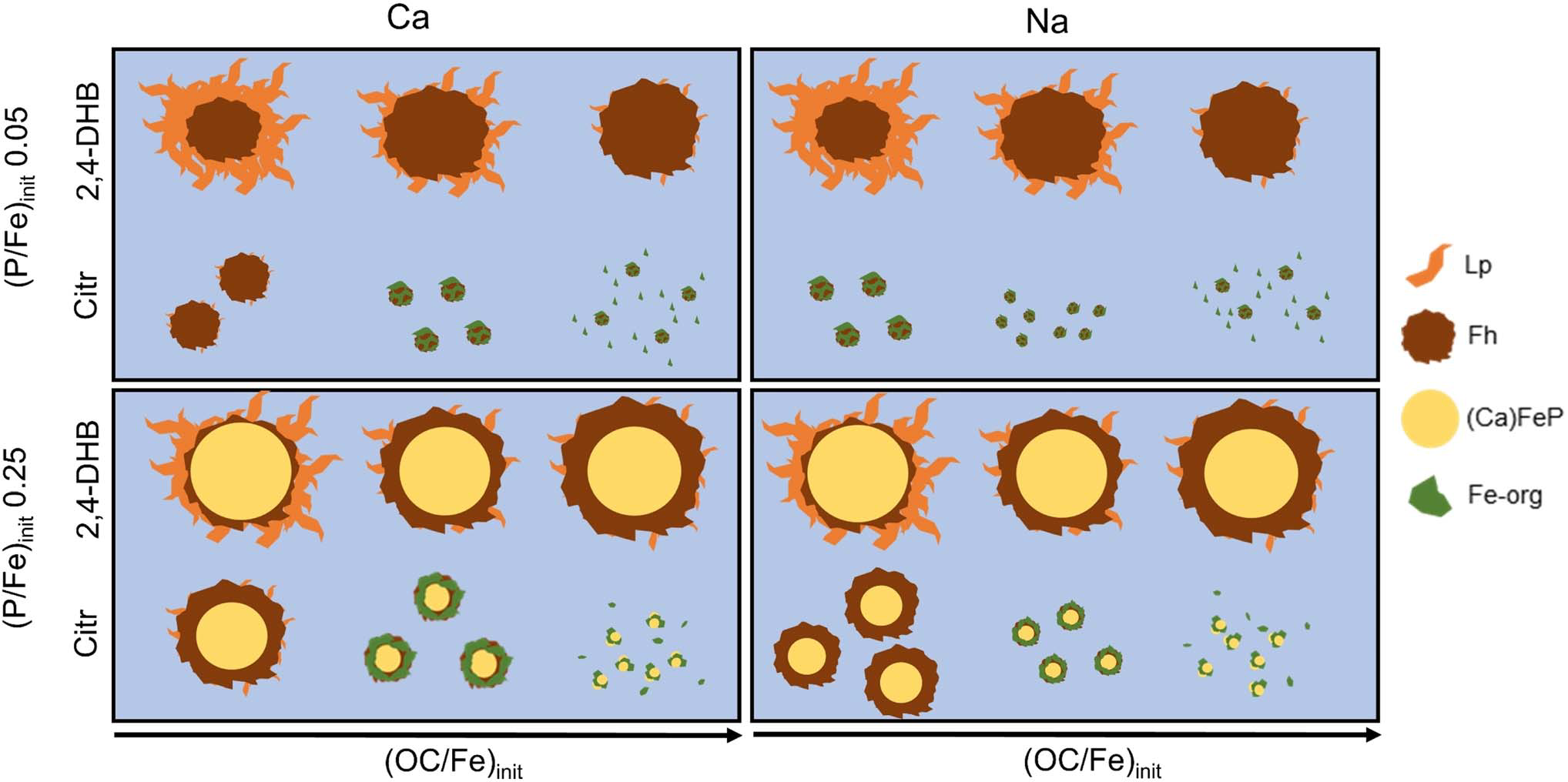
The researchers oxidized 0.5 mM Fe(II) in bicarbonate-buffered solutions (pH ≈ 7) to simulate natural water conditions. They tested four low-molecular-weight organic acids—2,4-dihydroxybenzoic acid (2,4-DHB), galacturonic acid (Galact), 3,4-dihydroxybenzoic acid (3,4-DHB), and citric acid (Citr)—as well as leonardite humic acid (LH) as a macromolecular analog of natural organic matter. These ligands span a gradient of Fe(III) complexation strength, from weak (2,4-DHB, Galact) to strong (3,4-DHB, Citr). The team systematically varied OC/Fe ratios (0.1–9.6), phosphate levels (PO₄/Fe = 0.05 and 0.25), and calcium concentrations (0 or 4 mM) to create 64 factorial combinations. Oxidation products were characterized using X-ray absorption spectroscopy (EXAFS), X-ray diffraction (XRD), Fourier-transform infrared spectroscopy (FTIR), and electron microscopy (STEM-EDX). Controls lacking organic matter formed amorphous Ca–Fe(III)–phosphate and ferrihydrite aggregates with lepidocrocite platelets attached, reflecting sequential Fe(III) mineralization. The introduction of organic ligands profoundly altered this structure. Weakly binding ligands (2,4-DHB, Galact) moderately suppressed lepidocrocite crystallization, whereas strong ligands (3,4-DHB, Citr) promoted amorphous, OC-rich ferrihydrite at the expense of crystalline phases. The EXAFS spectra confirmed reduced Fe–Fe coordination and increased monomeric Fe(III)–organic complexes with stronger or more abundant ligands. At low phosphate levels, organic ligands facilitated phosphate retention by stabilizing ferrihydrite surfaces with higher reactivity. However, at higher PO₄/Fe ratios, phosphate dominated Fe(III) coordination, diminishing organic co-precipitation. Interestingly, 3,4-DHB and Citr formed mixed (Ca)–Fe(III)–PO₄–OC nanoparticles that persisted as colloids, some smaller than 0.2 µm, indicating potential mobility through natural filtration barriers. Calcium intensified these interactions by promoting aggregation and co-precipitation of both phosphate and organic carbon. In contrast, leonardite humic acid produced broader, amorphous structures, suggesting steric hindrance rather than direct competition for Fe(III) sites.
Moreover, the authors found XRD patterns to show a progressive loss of lepidocrocite peaks with increasing ligand concentration and binding strength, replaced by broad ferrihydrite signals. FTIR analysis confirmed ligand-specific Fe coordination: catecholate-type binding for 3,4-DHB and carboxylate–hydroxyl coordination for Citr. Electron microscopy visualized nanoscale morphologies consistent with spectroscopic data—core–shell (Ca)FeP–ferrihydrite particles for weak ligands and dispersed, amorphous Fe–organic aggregates for strong ligands. Overall, the combination of EXAFS, XRD, and FTIR analyses established a consistent narrative: organic ligands, phosphate, and calcium co-regulate Fe(III) mineral formation, each shifting the equilibrium between crystallization, complexation, and aggregation.
In conclusion, the new findings of Professor Andreas Voegelin and colleagues redefine how we understand iron oxidation in natural systems. Rather than a simple competition between phosphate and organic carbon for Fe(III), the process emerges as a delicate balance of cooperative and antagonistic interactions. Weak organic ligands subtly delay mineral ordering but still allow phosphate-rich ferrihydrite and lepidocrocite to form, ensuring effective phosphorus immobilization. Stronger ligands, by contrast, create amorphous Fe–organic networks that trap phosphate and calcium into nanoscale colloids—entities likely to remain mobile in soils and aquatic interfaces. This duality suggests that organic composition and concentration dictate whether Fe-driven phosphorus retention stabilizes sediments or promotes its downstream transport. From an environmental perspective, the implications extend well beyond laboratory systems. The study demonstrates that iron’s role as both a sink and a shuttle for nutrients and carbon is conditional upon the organic milieu. In organic-rich environments such as wetlands, peatlands, or forest soils, high-affinity ligands like citrate or catechols could shift Fe(III) formation toward colloidal phases, reducing phosphorus sequestration but enhancing carbon preservation through complexation. Conversely, in mineral-dominated settings with low organic loads, ferrihydrite and lepidocrocite formation remains the dominant mechanism for phosphorus immobilization. Calcium’s synergistic role further emphasizes the coupling between geochemistry and biology. By promoting co-precipitation and aggregation, calcium not only reinforces phosphorus retention but also bridges organic macromolecules into denser clusters. This mechanism may help explain the stability of Fe–Ca–organic complexes in sediments and their resilience against reductive dissolution. The broader significance lies in linking nanoscale mineral formation to macroscopic biogeochemical cycles. The authors reveal that minor changes in ligand chemistry can modulate iron’s redox behavior, alter phosphorus fluxes, and control organic carbon turnover. Incorporating these parameters into environmental models could improve predictions of nutrient mobility under changing redox conditions and rising organic matter inputs from land use and climate change. Ultimately, the research highlights that Fe(III)-precipitate formation is not merely an inorganic process but a chemically dynamic intersection where organic and inorganic worlds meet. Understanding this balance offers a pathway to manage eutrophication, soil fertility, and carbon sequestration more effectively—anchoring iron chemistry as a key regulator of environmental resilience.


Reference
Nenonen, Ville & Kaegi, Ralf & Hug, Stephan & Luster, Jörg & Goettlicher, Joerg & Mangold, Stefan & Winkel, Lenny & Voegelin, Andreas. (2025). Effects of organic ligands, phosphate and Ca on the structure and composition of Fe(III)-precipitates formed by Fe(II) oxidation at near-neutral pH. Environmental science. Processes & impacts. 27. 10.1039/d4em00313f.
