
Significance
Excited electronic states in conjugated organic molecules often dissipate energy through intermolecular encounters once the molecules assemble into condensed phases. Planar polycyclic frameworks, which are frequently chosen for luminescent materials because they support extended π-conjugation and narrow optical transitions, tend to approach one another closely in the solid state. When that proximity occurs, excitons no longer remain confined to individual molecules. Aggregate formation, excimer emission, and exciton–exciton interactions begin to compete with radiative decay, broadening emission spectra and diverting energy into nonradiative channels. These effects become especially problematic in materials intended for deep-blue organic light-emitting diodes (OLEDs), where strict spectral requirements leave little tolerance for intermolecular perturbation. Multi-resonant thermally activated delayed fluorescence emitters address part of this problem through electronic design. Within these molecules, alternating electron-rich and electron-deficient atoms reshape frontier molecular orbitals so that the highest occupied and lowest unoccupied orbitals occupy different regions of the same framework. Spatial separation reduces exchange interaction between singlet and triplet excited states, narrowing the singlet–triplet energy gap. Thermal energy then enables triplet excitons to convert back into emissive singlet states, permitting OLED devices to harvest excitons that would otherwise decay without light emission. This mechanism has allowed organic emitters to reach impressive efficiencies while retaining spectrally sharp emission bands. The molecular skeletons that enable such electronic behavior introduce a separate structural liability. Multi-resonant emitters typically contain rigid, nearly planar polycyclic frameworks. That geometry encourages π-stacking and other intermolecular contacts once the molecules form thin films or crystalline phases. Even subtle intermolecular coupling can distort emission spectra or generate additional excited-state pathways. Color purity deteriorates, and radiative efficiency declines through new nonradiative processes. Efforts to mitigate these interactions have often relied on steric substitution. Bulky substituents can disrupt packing or reduce orbital overlap between neighboring emitters. Yet small positional variations in substituent placement frequently produce unpredictable outcomes in the solid state. Some derivatives maintain narrow emission bands, whereas closely related molecules still display spectral broadening or excimer formation. The relationship between steric modification and intermolecular suppression therefore remains difficult to control using conventional substituent strategies.
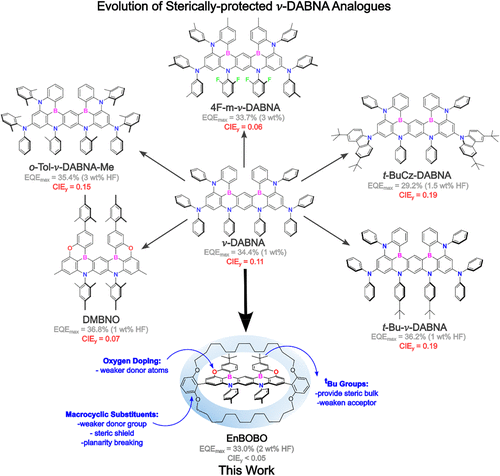
A different line of reasoning follows from a structural constraint imposed directly around the emissive core. Instead of decorating the periphery with isolated bulky groups, the molecular design explored here encloses the emitter within a covalently attached macrocycle. Such a structure introduces steric protection that extends above and below the molecular plane. If the macrocycle effectively restricts close intermolecular approach, the emissive core may retain the photophysical properties characteristic of isolated molecules even when embedded within a dense film. A recent research paper published in Journal of the American Chemical Society and conducted by Dr. Erin Holdsworth, Dr. Hwan-Hee Cho, Dr. Andrew Bond, Dr. Stephanie Montanaro, Dr. Seung-Je Woo, Dr. Tianyu Huang, Dr. Jordan Shaikh, Dr. Fathy Hassan, Dr. Sebastian Gorgon, Dr. Víctor Riesgo-Gonzalez, Dr. Alexander Gillett, Daniel Congrave and led by Professor Richard Friend and Professor Hugo Bronstein from the University of Cambridge, the authors developed a deep-blue multi-resonant thermally activated delayed fluorescence emitter in which a covalently attached macrocycle encloses the emissive core. The structure introduces steric separation that limits intermolecular aggregation and excimer formation in solid-state environments. Comparison with a non-encapsulated analogue demonstrates that the macrocycle preserves narrow emission spectra and improves photophysical efficiency. Integration into hyperfluorescent OLED devices yields high external quantum efficiency while maintaining deep-blue color coordinates. Briefly, the research team designed a molecular architecture derived from a boron–nitrogen doped multi-resonant framework related to ν-DABNA. They replaced peripheral diphenylamine groups with aryl substituents capable of bearing a macrocyclic ring that encases the emissive core. This substitution altered the electronic structure in a way that would normally shift emission toward longer wavelengths. The investigators compensated for that tendency by introducing oxygen atoms in place of selected amine donors and by adding tert-butyl substituents near the boron centers. These electronic adjustments widened the optical band gap so that the resulting emitter retained deep-blue photoluminescence despite the structural modification.
Synthetic work produced two related compounds. One molecule contained the full macrocyclic enclosure, while a second analogue retained the same emissive core but lacked the encapsulating alkyl chains. This pair allowed the researchers to evaluate how the macrocycle alters photophysical behavior without changing the central electronic structure. Nuclear magnetic resonance measurements revealed changes in alkyl proton environments after macrocyclization, consistent with restricted conformational motion of the chains surrounding the emitter. X-ray crystallography confirmed that the macrocycle positioned its alkyl segments above and below the planar core. Molecular packing analysis showed that adjacent emitters remained separated by distances exceeding those typical for π-stacking interactions, indicating that the ring structure effectively blocks close approach of neighboring cores. Solution-state spectroscopy demonstrated that both compounds retained the characteristic narrow emission associated with multi-resonant thermally activated delayed fluorescence materials. Absorption and emission spectra exhibited mirror-like symmetry consistent with transitions between the lowest singlet states. Emission maxima appeared in the deep-blue region, and spectral widths remained narrow, reflecting limited coupling between electronic and vibrational modes. The encapsulated emitter displayed slightly shorter emission wavelength and narrower spectral width compared with the open analogue, behavior consistent with reduced interaction between the excited-state dipole and the surrounding solvent environment.
The authors showed using transient photoluminescence measurements both prompt and delayed fluorescence components, confirming thermally activated delayed fluorescence activity. Quantitative analysis showed that the encapsulated molecule possessed a higher photoluminescence quantum yield and a faster radiative rate. At the same time, its intersystem crossing rate decreased relative to the open analogue. These changes suggest that the macrocycle subtly modifies the excited-state manifold, influencing how triplet and singlet states exchange population. Plus, device measurements explored how these molecular properties translate into electroluminescence. The investigators fabricated hyperfluorescent OLED architectures using the new emitters as terminal dopants in emissive layers containing thermally activated delayed fluorescence sensitizers. Devices incorporating the encapsulated emitter produced narrower and bluer electroluminescence spectra than those containing the non-encapsulated analogue. External quantum efficiencies approached the mid-thirty-percent range while maintaining deep-blue color coordinates compatible with stringent display standards. Additionally, using time-resolved spectroscopy on device films provided further clarification. The research group observed minimal spectral evolution in films containing the encapsulated emitter, indicating that emission arises largely from a single molecular species. Films containing the non-encapsulated analogue developed additional red-shifted emission components over time, consistent with aggregate or excimer formation. The contrast between the two systems reveals a direct link between the macrocycle’s steric constraint and suppression of intermolecular excited-state interactions.
To summarize, Professor Richard Friend and Professor Hugo Bronstein and colleagues demonstrated that deep-blue OLED emitters must satisfy two difficult requirements simultaneously. They must produce spectrally narrow emission centered at short wavelengths while maintaining high electroluminescent efficiency. These objectives conflict because the planar conjugated structures that yield sharp optical transitions tend to aggregate in thin films. Once molecules interact strongly with their neighbors, spectral purity deteriorates and nonradiative decay channels emerge. The molecular architecture explored in this study reframes that design challenge. Encapsulation imposes a structural barrier that physically separates emissive cores without altering their fundamental electronic topology. In practical terms, the macrocycle functions as a three-dimensional steric shield. By extending above and below the planar chromophore, the ring prevents neighboring molecules from approaching closely enough to form excimers or strongly coupled aggregates. A second effect emerges from the same structural constraint. The macrocycle partially isolates the excited-state dipole of the emitter from its surrounding medium. Solution experiments revealed reduced sensitivity of the encapsulated molecule to solvent polarity. This observation carries practical consequences for device operation, since the dielectric environment of an OLED film differs markedly from dilute solution. If the emissive core experiences less environmental perturbation, spectral shifts and line broadening diminish.
These two structural consequences—suppression of intermolecular contact and insulation from environmental polarity—combine to stabilize the excited-state dynamics of the emitter. Radiative decay remains efficient, and delayed fluorescence continues to harvest triplet excitons without introducing strong spectral distortion. Device measurements demonstrate that this stability translates into electroluminescent performance approaching the limits expected for organic emitters while preserving deep-blue emission coordinates. The work of University of Cambridge scientists also highlights a methodological point. Time-resolved photoluminescence proved necessary to detect weak emissive species associated with aggregation. In steady-state measurements those signals contribute little intensity and remain obscured by the dominant emission band. Temporal resolution reveals their formation and evolution, allowing the role of intermolecular processes to be identified directly. Encapsulation introduces a synthetic direction that differs from incremental substitution strategies. Instead of adjusting steric bulk through discrete substituents, the macrocycle enforces a spatial constraint that operates continuously across the entire chromophore. Materials scientists seeking to control excited-state interactions in densely packed organic films may find this architectural approach adaptable to other emissive systems.


Reference
Holdsworth EM, Cho HH, Bond AD, Montanaro S, Woo SJ, Huang T, Shaikh J, Hassan F, Gorgon S, Riesgo-Gonzalez V, Gillett AJ, Congrave DG, Friend RH, Bronstein HA. Macrocyclic Covalent Encapsulation of a Multi-Resonant Emitter: Understanding and Controlling Interactions in Highly Efficient Deep-Blue OLEDs. J Am Chem Soc. 2026 Mar 4;148(8):8163-8173. doi: 10.1021/jacs.5c16290.