Significance
Technological advancement in the field of organic electronics has led to the development of high-performance devices with applications in numerous areas including displays and lighting. Generally, the performance of these devices depends on the oxidation potentials (EOX) and reduction potentials (ERED) of the materials from which they are made. Unfortunately, a number of studies have shown that it is always difficult to determine the reduction potential and oxidation potential of these materials experimentally owing to several factors such as poor compound solubility and potential values exceeding the anticipated range.
Considering the future trends, the need for high performing organic electronic devises has significantly increased thus highly efficient methods for determining redox potentials in organic semiconductors are highly desirable. Alternatively, among the available methods for determining the redox potentials, density functional theory is widely preferred due to their reliability and low cost. Despite the great achievements, studies involving predicting reduction potential and oxidation potential for materials used in developing organic light emitting diodes are still missing.
To this note, Zhejiang University researchers: Dr. Dan Wang, Dr. Chao Wang, MissYan Yue and Professor Qisheng Zhang from Department of polymer science and Engineering in collaboration with Professor Shuping Huang at Fuzhou University cross-examined the oxidation and reduction potentials of 50 different compounds. In particular, they purposed to develop a general method for determining the redox potentials for different organic compounds and especially those used in light emitting diodes. Their research work is currently published in the research journal, Organic Electronics.
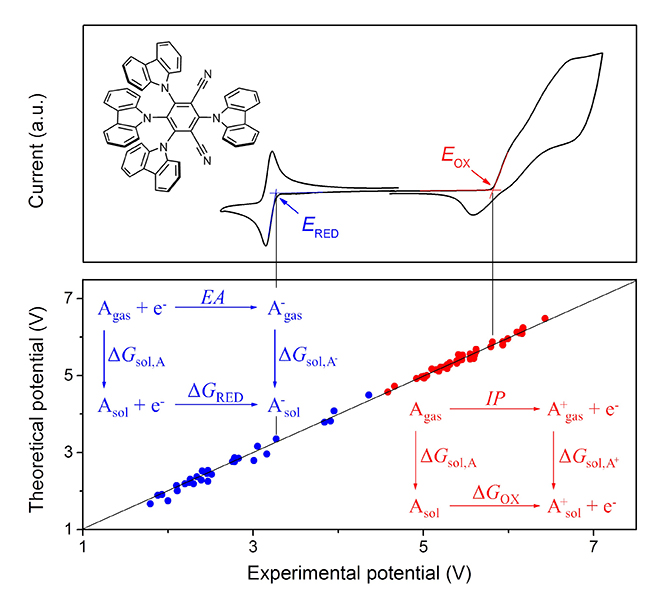
In brief, the research team commenced their research work by evaluating the performance of 13 different density functions such as PBE and M06. The reduction and oxidation potentials were reproduced in acetonitrile and dichloromethane solutions, respectively, on the basis of thermodynamic cycles for electron transfer. The researchers then investigated the relationship and influence of the Hartree-Fock exchange on both the oxidation and reduction potential. To actualize their study, the results obtained were compared to those obtained experimentally through cyclic voltammetry.
From the conducted experiments, the authors observed that the generalized developed method produced oxidation and reduction potentials with a corresponding mean absolute error of 0.05 V and 0.08 V, respectively. In addition, Hartree-Fock exchange played significant roles which varied from one density functional group to another like in the cases demonstrated by the GGA and hybrid GGA functionals. Furthermore, the oxidation potential increased while the reduction potential decreased with the increase of the Hartree-Fock exchange percentage.
In summary, the Zhejiang University scientists were the first to accurately calculate the oxidation and reduction potentials for a large variety of small molecules based on the density functional theory methods. A rough linear relationship between the highest occupied molecular orbital energies and theoretical oxidation potential and as well as lowest unoccupied molecular orbital energies and theoretical reduction potential was also revealed. Owing to the accuracy and low cost of the method, it will pave way for accurate prediction of redox potentials for materials used in organic light emitting diodes thus will advance lighting and display applications.
Reference
Wang, D., Huang, S., Wang, C., Yue, Y., & Zhang, Q. (2019). Computational prediction for oxidation and reduction potentials of organic molecules used in organic light-emitting diodes. Organic Electronics, 64, 216-222.
Go To Organic Electronics