Significance
Phase-change materials (PCMs) have emerged as one of the most promising candidates for non-volatile memory technologies. Unlike traditional volatile memory (e.g., DRAM), non-volatile memory retains data even when power is turned off, making it ideal for applications requiring persistent data storage. PCMs, in particular, exhibit a remarkable property: they can swiftly and reversibly transition between amorphous and crystalline phases through processes known as crystallization (SET) and amorphization (RESET). This property allows for the encoding of digital information by exploiting substantial contrasts in physical properties between the amorphous and crystalline phases, such as electrical conductivity and optical reflectivity. Among the PCMs studied extensively, those located on the GeTe–Sb2Te3 (GST) quasi-binary line have garnered significant attention. Variants of GST, including Ge2Sb2Te5 and Ge1Sb2Te4, have been used in various applications, from optical discs to cross-point random-access memory products. Additionally, PCMs based on Sb2Te3 with suitable dopants or nano-confinement layers show great promise for universal memory and neuro-inspired computing devices. The versatility of GST-based PCMs extends to on-chip non-volatile photonic applications, further expanding their potential impact. Understanding the intricate behavior of PCMs at the atomic level is essential for optimizing their use in practical devices. Density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations have been valuable tools for gaining insights into the structural and chemical aspects of GST-based PCMs, including their crystallization kinetics. However, AIMD simulations have limitations, as they are computationally expensive and restricted to systems with a limited number of atoms and short timescales. Additionally, the complexity of PCMs necessitates quantum-mechanically accurate methods rather than empirically parameterized force-field models. Furthermore, simulating realistic scenarios encountered in devices, such as temperature gradients due to Joule heating, volume changes during phase transitions, and local chemical composition evolution, requires larger model system sizes on the order of tens of nanometers.
A new study, led by Professor Volker Deringer and his team at the University of Oxford’s Department of Chemistry, Inorganic Chemistry Laboratory, sheds light on this fascinating field by harnessing the power of machine learning (ML) to conduct atomistic simulations of PCMs along the GeTe–Sb2Te3 compositional line. This pioneering work, published in the peer-reviewed journal Nature Electronics, promises to accelerate the development of novel memory devices and computing architectures based on PCMs.
Machine learning-based interatomic potential models have emerged as a promising approach to bridge the gap between computational efficiency and quantum-mechanical accuracy. These models combine the speed of empirical potentials with the accuracy of DFT. In this context, the team led by Professor Deringer demonstrated the potential of machine learning-driven modeling to perform fully atomistic device-scale simulations of phase changes along the GST compositional line, even under realistic device geometries and conditions. Their approach involved the development of an artificial neural network machine learning model using the Gaussian approximation potential (GAP) framework. The model was trained on a comprehensive reference database of structural configurations and DFT-computed energies and forces. Importantly, the training process iteratively refined the model’s accuracy, allowing it to encompass a wide range of structural scenarios relevant to GST PCMs. This database-driven machine learning approach resulted in a robust ML model, referred to as GST-GAP-22, capable of predicting structural properties with high fidelity.
The GST-GAP-22 model demonstrated remarkable predictive power. It accurately reproduced the structural properties of crystalline phases, including lattice parameters, and successfully captured the radial, angular, and ring-size distributions of liquid and amorphous GST phases. Moreover, it accurately represented the fraction of homopolar bonds in amorphous GST, a significant factor in aging and resistance drift. The model also captured the Peierls distortion effect, crucial for understanding bandgap properties in crystalline and amorphous GST.
Beyond the GST quasi-binary line, the model showed transferability to slightly off-stoichiometric compositions, including p-type and n-type semiconducting behavior. This compositionally transferable and defect-tolerant nature of the ML model enabled the efficient simulation of amorphous GST with local compositional fluctuations, mimicking real-world device conditions. The ML-driven simulations enabled the study of complex phase-change processes in PCMs, such as crystallization (SET) and amorphization (RESET). These simulations provided atomistic insights into the mechanisms of these processes, shedding light on the structural changes that occur during phase transitions. Significantly, these simulations allowed for the emulation of cumulative SET processes, a critical aspect of neuro-inspired computing, by varying pulse amplitudes and durations. Such simulations hold the potential to mimic synaptic learning rules and contribute to the development of stochastic phase-change neurons.
One of the most impressive achievements of this research was the ability to surpass the computational limitations of AIMD simulations. While AIMD simulations are limited to small-scale systems and short timescales, the ML-driven simulations demonstrated the potential to model large-scale crystallization, even in nanoscale devices. These simulations offered valuable insights into polycrystalline growth, providing a bridge between small-scale models and real-device-size simulations.
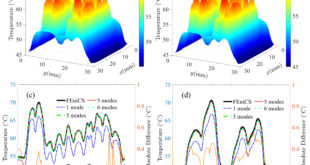
Moving closer to practical applications, the research team conducted simulations at the device scale, specifically modeling a cross-point memory device. This device-scale simulation involved modeling a PCM memory layer within the context of a larger device, complete with buffer layers and dielectric materials. The simulation accurately reproduced the progressive disordering and amorphization of the PCM region in response to heating, mirroring real-world programming conditions. The results provided a glimpse into the complex thermal dynamics that occur during device operation, including heat dissipation and phase transitions.
The research team also demonstrated the potential of their ML-driven simulations to address electromigration, a critical consideration in PCM devices. By applying an electric field, they accelerated the migration of ions within the PCM material, providing insights into potential failure mechanisms. This aspect of the research showcases the versatility of ML-driven simulations in addressing various device-related challenges.
The work presented in this study holds immense promise for advancing the field of phase-change materials and their applications in non-volatile memory and neuro-inspired computing technologies. ML-driven simulations, as demonstrated by Professor Deringer’s team, offer a powerful tool for exploring the behavior of PCMs at the atomic level, bridging the gap between theory and practical device engineering. Future research in this area may focus on further extending the ML models to capture interface effects between PCM materials and surrounding layers, including electrodes and dielectrics. Additionally, simulations can be tailored to investigate a broader range of programming conditions, such as varying pulse amplitudes, durations, and shapes, to optimize device performance. The combination of ML potentials with electric-field effects opens new avenues for realistic modeling of PCM devices and their long-term reliability.
In conclusion, the study led by Professor Volker Deringer represents a significant leap forward in our understanding and modeling of phase-change materials. By harnessing the power of machine learning, the research team has demonstrated the capability to perform atomistic simulations of PCMs at various scales, from small-scale crystallization to device-level modeling. These simulations provide invaluable insights into the behavior of PCMs under realistic conditions, paving the way for the development of next-generation non-volatile memory devices and neuro-inspired computing architectures. The interdisciplinary approach showcased in this research, merging materials science, machine learning, and engineering, holds great promise for the future of information technology. As we continue to push the boundaries of our understanding of materials at the atomic level, we can look forward to transformative innovations in memory technology and beyond.


Reference
Yuxing Zhou, Wei Zhang, En Ma & Volker L. Deringer. Device-scale atomistic modelling of phase-change memory materials. Nature Electronics volume 6, pages746–754 (2023)