Significance
Among the established smart macromolecular containers, one stands out. Layered double hydroxides generally exhibit bifunctional capacity with the ability to host functional molecules. Generally, they have numerous beneficial uses in different areas including drug delivery, adsorption, and catalysis. This is however determined by the nature of the anions that constitutes the doubled hydroxides.
Specifically, in applications such as corrosion, layered double hydroxides act as reservoirs for functional anions due to their anion-exchange characteristics, a step that has significantly enhanced their corrosion protection capabilities. Unfortunately, exploring and developing different applications of layered double hydroxides involving ion exchange and equilibrium processes between their interlayers has remained a great challenge. This has been attributed to the difficulty in performing accurate molecular dynamics simulations to analyze the dependence of various layered double hydroxides applications on the equilibrium mechanisms and ion exchange.
To this note, a team of researchers at the University of Aveiro in Portugal: Dr. Germán Pérez-Sánchez and Dr. José Gomes, from the Department of Chemistry, in collaboration with Dr. Tiago Galvão and Dr. João Tedim, from the Department of Materials and Ceramics Engineering, developed a computer simulation model to investigate the role of the various intercalated anions in the basal space. In the framework of project SELMA (Unveiling the self-healing mechanisms associated with smart nanocontainers, PTDC/QEQ-QFI/4719/2014, financed by national funds through FCT/MEC (PIDDAC) and co-financed by FEDER under the PT2020 Partnership Agreement), they aimed at modeling different applications of layered double hydroxides involving interlayer equilibrium processes and ion-exchange. In particular, layered double hydroxides constituting zinc-aluminum and magnesium-aluminum with a metallic ratio of 2:1 were used to validate the computer model taking into consideration the initially published work. Their research work is currently published in the journal, Applied Clay Science.
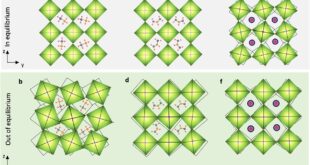
Briefly, the research method employed entailed a cross-examination of the various applications of layered double hydroxides that involves ion exchange and interlayer equilibrium process. Firstly, atomic partial charges, obtained using the DDEC6 formalism and electron distributions computed with a computational approach based on the Density Functional Theory (DFT calculations, were incorporated with the existing parameters in the literature to model the Mg2Al LDH with different anions. In particular, the model accounted for the periodic expansion of layered double hydroxides structure in parallel and perpendicular directions to cationic layers with different anions. Eventually, a comparison was conducted between the calculated results and experimental data to verify the functionality of the developed model.
The Portuguese research team observed that the proposed computer model was effective for long molecular dynamics simulations without compromising the integrity of the layered double hydroxides while at the same time allowing for free movement of the atomic positions. Additionally, it was also noted that the materials with nitrate anions exhibited larger interlayer distances as compared to those with carbonate anions. The larger charge density in the latter leads the carbonate anions to arrange parallel to the LDH metal layer plane conversely to the tilt positions observed in nitrate anions. The computer model was able to mimic the experimental conditions in which the LDH is completely immersed in a corrosive chloride aqueous solution.
In summary, the study by the University of Aveiro scientists successfully demonstrated a new molecular dynamics approach for investigating the structure and dynamics of layered double hydroxides. Interestingly, despite the long simulation time, the LDH maintained the structure with different ions and the LDH in bulk solution and the initial stages of the nitrate-chloride ion exchange could be observed. Altogether, the study will pave the way for analyzing water or ion exchange between the layered double hydroxides and solutions, a great step in enhancing different applications involving ion-exchange and interlayer equilibrium processes.




Reference
Pérez-Sánchez, G., Galvão, T., Tedim, J., & Gomes, J. (2018). A molecular dynamics framework to explore the structure and dynamics of layered double hydroxides. Applied Clay Science, 163, 164-177.
Go To Applied Clay Science