
SIGNIFICANCE
The promise of hydrogen as a clean fuel is genuine, but the metal–hydrogen relationship has always been complicated. A single hydrogen atom, tiny enough to slip almost anywhere and reactive enough to disturb local bonding, can quietly change how steel carries load. In practice, that means toughness can fall, and fracture can initiate far earlier than expected. Scientists have offered competing explanations for this behaviour over the years, however, most agree on the early steps. Hydrogen must first dissociate at the surface, then adsorb, and eventually settle into microstructural imperfections. Once it does, the risk of embrittlement becomes very real. Grain boundaries tend to draw special attention because they do not behave like the surrounding lattice. Their geometry and chemistry create small pockets of altered electronic structure, which can either act as barriers or serve as very comfortable stopping points for hydrogen. That duality makes them intriguing and frustrating at the same time.
Hydrogen molecules breaking apart on clean Fe surfaces and atoms diffusing inward come mostly from idealized models, but actual pipeline steels are messier because they contain alloying elements added for strength or corrosion resistance, and these elements often migrate toward grain boundaries. Their presence changes the local stress state and electron distribution in ways that are not always predictable, which leaves us with a practical question: do these solutes help protect the boundary, or do they accidentally make hydrogen uptake worse? Experimental data at the pressures relevant to pipelines are scarce. To this end, a new research paper published in Energy & Fuels, conducted by Aliakbar Sheikhzadeh, Associate Professor Jing Liu and led by Professor Hao Zhang from the Department of Chemical and Materials Engineering at the University of Alberta, in collaboration with Dr. Yimin Zeng from Natural Resources Canada, researchers developed two complementary first-principles models: one capturing hydrogen dissociation and adsorption at surface Σ5(310)/[001] grain boundaries and another describing hydrogen solution behavior within bulk grain boundaries. These models systematically evaluate alloy segregation, electronic structure modifications, and adsorption energetics across ten solute elements.
The research team constructed a Σ5(310)/[001] Fe grain boundary using a four-layer Fe(100) slab and examined hydrogen behavior through density-functional theory (DFT) calculations implemented in VASP. Alloying elements (Al, Cr, Cu, Co, Mn, Mo, Ni, Ti, V, and W) were introduced at substitutional sites either at the free surface or within the bulk region of the grain boundary. For each alloying concentration, the team evaluated up to nineteen possible configurations to identify energetically stable solute arrangements before introducing hydrogen for dissociation or adsorption analysis, as represented in Figure 1. The authors showed that hydrogen molecules inevitably dissociate at the alloyed grain boundary surface regardless of the dopant. They reported the dissociative adsorption energies, which revealed that even elements previously shown to suppress H–H bond cleavage on flat Fe(100) surfaces—such as Al or Cu—cannot prevent dissociation within the grain-boundary environment. The team performed projected density-of-states plots and differential charge-density maps, which illustrated how the boundary itself amplifies orbital hybridization, enabling strong interaction between Fe d-states and hydrogen 1s orbitals. In some cases, such as W and Co, hydrogen interacts more strongly with the dopant than with surrounding Fe atoms, reflecting a localized electronic environment that deepens the adsorption well. When the team examined atomic hydrogen adsorption at the surface grain boundary, alloy-dependent trends became clearer, and energy amounts are represented in Figure 2. Elements such as Co, Mn, Ni, Ti, and W consistently produced more negative adsorption energies, often exceeding −1.0 eV. Charge-density maps show that Ti increases electron donation toward hydrogen, enhancing its trapping. By contrast, Al, Cr, Cu, Mo, and V exhibit weaker trapping strengths, and their ability to bind hydrogen diminishes as their concentration increases. In these cases, dopant clusters appear to reduce local charge transfer, making the adsorption energetically less favorable. The authors reported that the situation to change markedly in the bulk grain boundary and all alloying elements show strong segregation tendencies, but hydrogen becomes less stable as solute concentration increases, as shown in Figure 2. Additionally, solution energies for hydrogen become positive in several cases, especially at high concentrations of Mo, Al, and W. Voronoi volume analyses and strain maps reveal that accumulated dopants impose compressive strain that shrinks interstitial free volume. Under these conditions, the bulk boundary does not behave as a rapid hydrogen-diffusion pathway but instead becomes energetically inhospitable to hydrogen.
|
1 Alloy |
|
|
2 Alloys |
|
|
3 Alloys |
|
|
4 Alloys |
|
|
5 Alloys |
|
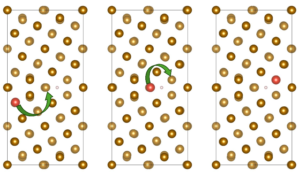
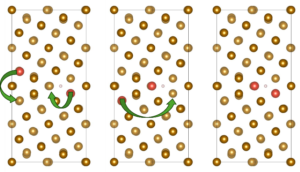
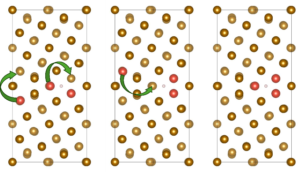
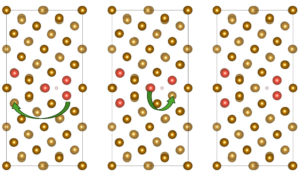
Figure 1: Configurations of ∑5(310)/[001] grain boundary in the presence of doping elements with different concentrations. The yellow color represents Fe atoms and the red color represents the doping element atoms.
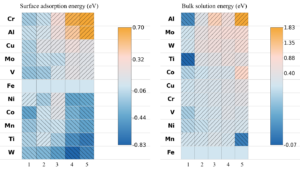
Figure 2: Surface adsorption energy and bulk solution energy for various alloyed grain boundaries. Each contour map displays the number of alloying atoms along the X-axis and the type of alloying element along the Y-axis.
In conclusion, Professor Hao Zhang and colleagues demonstrated how specific dopants either enhance surface trapping or suppress bulk solubility through strain-induced confinement. The new dual-model framework provided a predictive route to designing alloy chemistries that mitigate hydrogen uptake in steels. An important implication of the new work is that grain boundaries doped with certain elements, notably Co, Mn, Ni, Ti, and W, develop strong surface trapping characteristics. Because these dopants deepen the adsorption potential wells at the surface, atomic hydrogen becomes more tightly bound there, reducing the likelihood of penetrating deeper into the steel, as represented in Figure 2. Moreover, the PDOS and charge-density evidence support this interpretation, and show strengthened orbital interactions at alloyed surface sites. For steels intended for high-pressure hydrogen service, this suggests a possible alloy-design strategy: promoting solute segregation at exposed boundaries could decelerate hydrogen ingress by immobilizing hydrogen at the surface.
We believe equally important is the University of Alberta scientists’ finding that the bulk grain boundaries behave differently. While solutes readily segregate to these boundaries, their accumulation compresses the surrounding lattice, diminishing accessible free volume for hydrogen. The positive solution energies at high solute concentrations indicate that, beyond a certain threshold, hydrogen is effectively repelled, which challenges the common assumption that grain boundaries facilitate rapid long-range hydrogen migration. Instead, depending on their solute chemistry and segregation density, grain boundaries may serve as bottlenecks rather than highways for hydrogen diffusion. From a metallurgical standpoint, this dichotomy between surface and bulk behavior matters. Pipeline steels will inevitably experience dynamic environmental conditions in which hydrogen must first dissociate at the outermost surface before attempting to diffuse inward. If alloy-segregated boundaries at the surface function as strong traps, they may act as protective layers, similar in spirit to tailored oxide films or engineered diffusion barriers. If excessive solute clustering occurs in the bulk, the resulting compressive strain fields may reduce hydrogen solubility but also modify mechanical stability. Careful alloy-design strategies must therefore consider both beneficial hydrogen-blocking effects and potential side effects due to local lattice distortion. In a nutshell, engineers and scientists who design steels for hydrogen pipelines, storage vessels, or compressors can use the new results of Professor Hao Zhang and colleagues to screen alloying additions that either promote safe hydrogen retention at the surface or limit bulk uptake.

Reference
Sheikhzadeh, Aliakbar & Liu, Jing & Zeng, Yimin & Zhang, Hao. (2025). First-Principles Evaluation of Alloying Effects on Hydrogen Adsorption and Trapping at Grain Boundaries in Pipeline Steels. Energy & Fuels. 39. 15210-15223. 10.1021/acs.energyfuels.5c02772.
