Significance
Elasticity is a fundamental property of materials, enabling a reversible response to applied stress, which is critical for both biological functions and engineering applications. Despite its ubiquity, the precise atomic-scale mechanisms responsible for elasticity in molecular crystals remain largely unresolved. This gap in understanding limits the ability to design advanced materials with tailored mechanical properties. A major challenge in this field is identifying the specific intermolecular interactions that govern elasticity. Traditional models treat elasticity as a bulk material property, assuming that multiple interactions collectively contribute to the restoring force. However, in molecular crystals, where covalent bonds remain largely undeformed under elastic stress, weak intermolecular interactions dictate mechanical behavior. The fundamental question that remains unanswered is where within the crystal the restoring force originates and how different types of interactions contribute to it. Previous studies have established that molecular flexibility in crystalline materials is often anisotropic, meaning that different crystallographic directions exhibit varying degrees of elasticity. However, pinpointing the interactions responsible for restoring elasticity at the atomic scale has proven difficult due to the complexity of crystal packing and the multitude of forces at play. Recent advances in structural determination techniques, such as high-resolution diffraction studies of elastically deformed crystals, now allow for the precise mapping of these forces.
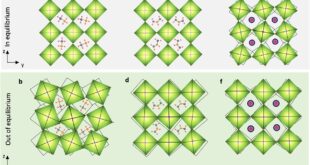
New research paper published in Journal of Nature Materials and conducted by Dr. Amy Thompson, Dr. Bowie Chong, Dr. Elise Kenny, Dr. Jack Evans, Dr. Joshua Powell, Dr. Mark Spackman, Professor John McMurtrie, Professor Benjamin Powell & led by Professor Jack Clegg from The University of Queensland, Brisbane, Queensland in Australia, the researchers employed a series of density functional theory (DFT) calculations on single crystals of three structurally related molecular materials: bis(acetylacetonato)copper(II) ([Cu(acac)2], denoted as [CuL1₂]), along with two derivatives featuring methyl-to-ethyl and methyl-to-propyl substitutions. The study aimed to determine how different intermolecular interactions influence elasticity under both compressive and expansive strain. By analyzing these materials, the research seeks to reveal the atomic-level mechanisms underlying elastic behavior, ultimately facilitating the rational design of flexible molecular materials for applications in photonic circuits, optical waveguides, and other advanced technologies. The authors examined three key intermolecular interactions within [CuL1₂]: intrachain π-π stacking, CH-π interactions, and CH-O interactions. These forces were quantified using energy framework calculations to determine their relative contributions to elasticity. The experimental methodology involved subjecting single crystals of [CuL1₂] to controlled elastic bending and analyzing structural changes through microfocused X-ray diffraction. The findings revealed that the elasticity in [CuL1₂] arises from distinct interactions depending on whether the crystal is under expansive or compressive strain. Under expansive strain, the restoring force originates primarily from π-π interactions, while under compressive strain, CH-π and CH-O interactions play a dominant role.
Energy calculations showed that individual interactions within [CuL1₂] exhibit their energy minima outside the observed elastic limit of the crystal, meaning that the overall elastic response results from a combination of frustrated interactions. When bent, the sum of these interactions reproduces the equilibrium crystal structure, confirming their collective role in elasticity. The introduction of larger alkyl groups (ethyl and propyl) in [CuL2₂] and [CuL3₂] altered the balance of these interactions, leading to variations in mechanical flexibility. Specifically, [CuL3₂] exhibited a much stiffer response due to an increase in repulsive CH-HC interactions. Additionally, the researchers found that the potential energy stored in an elastically bent [CuL1₂] crystal corresponds to approximately 90 J mol⁻¹, which is comparable to many mechanical energy scales. In practical terms, this stored energy could theoretically lift an object 30 times the crystal’s own weight by 1 meter against gravity. This mechanical potential was experimentally demonstrated by attaching steel spheres to bent single-crystal cantilevers, showing that the stored elastic energy could be used to raise the spheres upon release.
In conclusion, Professor Jack Clegg and colleagues demonstrated the molecular origins of elasticity in crystalline materials and identified the atomic-scale interactions responsible for elastic restoring forces, the research offers a new framework for designing mechanically tunable materials. This understanding has significant implications for the development of flexible optical components, wearable electronics, and adaptive materials that require precise mechanical responses. They also demonstrated that elasticity in molecular crystals is governed by specific interactions rather than being a bulk material property. This suggests that by modifying molecular packing and intermolecular forces, researchers can engineer materials with customizable elastic properties. The results also highlight the role of interaction frustration in defining mechanical behavior, a concept that may extend to other fields such as quantum magnetism and spin-crossover materials. Potential applications of this research include the design of mechanically flexible materials for photonic circuits, where controlled elastic deformation can influence optical properties. The identification of distinct restoring forces under expansive and compressive strain also informs the development of advanced structural materials, such as lightweight, impact-resistant components.




Reference
Thompson, A.J., Chong, B.S.K., Kenny, E.P. et al. Origins of elasticity in molecular materials. Nat. Mater. (2025). https://doi.org/10.1038/s41563-025-02133-w