
Significance
Molecular solvation in light of the interaction of a salute and solvent molecules has been found to be significant in understand the dynamic, spectroscopic, and thermodynamic features of materials. Implicit solvation methods, which approximate the solvent as a dielectric continuum, have been adopted in defining the solvation free energy as well as the derived thermodynamic features of mixed and pure fluids.
In the case of solvation methods, the dielectric response of a solvent reference to the presence of a solute can be obtained from the apparent charges at the solute-solvent boundary. In case the solvent is a perfect conductor, it is easier to obtain screening charges. This is in view of the fact that at the solute-solvent boundary, the net electric potential is zero.
COSMO-based activity coefficient models provide accurate predictions for an array of thermodynamic features and phase equilibria such as liquid-liquid, vapor-liquid, and solid-liquid equilibria for mixtures of electrolytes, water, ionic liquids, and organics. Unfortunately, despite these huge steps made by researchers, COSMO-based models are still less accurate for associating fluids. Hydrogen bonding interactions have been considered to occur between molecular surfaces related to high and opposite screening charge densities. However, the integrity of a hydrogen bond should be quantified by the screening charge density and an empirical interaction constant whose value is established to reproduce experimental phase equilibrium data.
Efforts have been made to enhance the prediction accuracy of COSMO-based approaches by introducing temperature-dependent interaction and donor-acceptor dependent interaction parameters. These efforts have enhanced the definition of associating fluids, but this success has been realized at the expense of introducing empirical parameters.
Professor Shiang-Tai Lin and his PhD student Wei-Lin Chen at the National Taiwan University developed a new model taking into account the partial orientational constraints in hydrogen bonds for predicting the phase equilibrium of mixtures implementing the COSMO-SAC model. Their research work is published in journal, Physical Chemistry Chemical Physics.
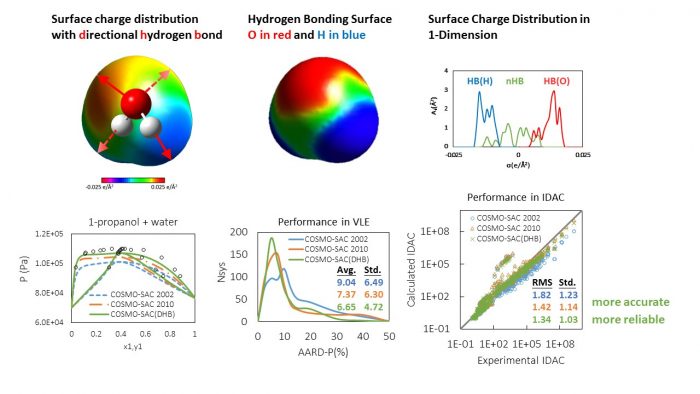
The already existing approaches for defining the hydrogen bonding interaction consider the integrity of the interaction based solely on screening charges polarity, yet neglecting the finding that hydrogen bond formation calls for particular orientation between the acceptor and donor pairs.
In the new approach, denoted as COSMO-SAC(DHB), the authors recognized the hydrogen bond segments within the area of the donor-acceptor pair. They identified the spatial locations for the allowed hydrogen bond segment based on molecular orientation and Valence Shell Electron Pair Repulsion theory.
The proposed model by Lin and Chen required only three universal parameters to define the dimensions of the contacting surface area of the donor-acceptor pairs, the integrity of the hydrogen bond interaction, and the cutoff charge density for the formation of a hydrogen bond. Counting on an analysis of a large data base of liquid–liquid equilibrium, vapor–liquid equilibria, infinite dilution activity coefficient, and water-octanol partition coefficient, the authors concluded that the incorporation of the directional constraints was effective in enhancing the precision and reliability of the COSMO-based methods.


Reference
Wei-Lin Chen and Shiang-Tai Lin. Explicit consideration of spatial hydrogen bonding direction for activity coefficient prediction based on implicit solvation calculations. Physical Chemistry Chemical Physics, volume (2017), 20367
Go To Physical Chemistry Chemical Physics
