Significance Statement
Graphene is a remarkable material with outstanding properties, and many applications have been developed based on this material. For example, freestanding graphene monolayers can be mounted on small apertures to create pressure sensors as well as in making freestanding devices and resonators. Thin films of graphene can also be used to protect surfaces. Graphene is already being used to create bendable displays for potential use in cell phones. Graphene has been predicted to have a melting point of 4500 K. Due to the high melting point of graphene and carbon nanotubes, further investigation of their high temperature properties is desirable.
Because of its high thermal stability, graphene can be successfully applied in high temperature electronic gadgets. Recently, a light-bulb was fabricated using a filament made of reduced graphene oxide together with single walled nanotubes. The reduced graphene oxide single-walled nanotubes can reach about 3000K, with some samples even reaching 3300K before failure.
Given the high melting points of graphene and analogues such as carbon nanotubes, and in light of their potential high temperature applications, it is very important to have in-depth and accurate information on the initial stages of graphene melting. Therefore, a team of researchers under the guidance of Prof. Eric Ganz at the University of Minnesota in collaboration with Prof. Li-Ming Yang at Huazhong University of Science and Technology, and Ariel B. Ganz at Cornell University used an accurate ab initio density functional theory molecular dynamics simulations to analyze the initial stages of melting of freestanding graphene at temperatures ranging from 4000-6000K. Their work has now been published in Physical Chemistry Chemical Physics.
The authors performed ab initio density functional theory molecular dynamics simulations using a 10 x 10 graphene lattice with periodic boundary conditions. At 0 K the graphene unit cell would be 24.6 x 24.6 x 20.0 Å, therefore, the authors used a slightly larger unit cell in order to accommodate expansion as the temperature rose. The 4000 K simulation started from an ideal 0K graphene lattice. For every simulation, approximately 12 ps equilibration time was used. The researchers collected the data after equilibration.
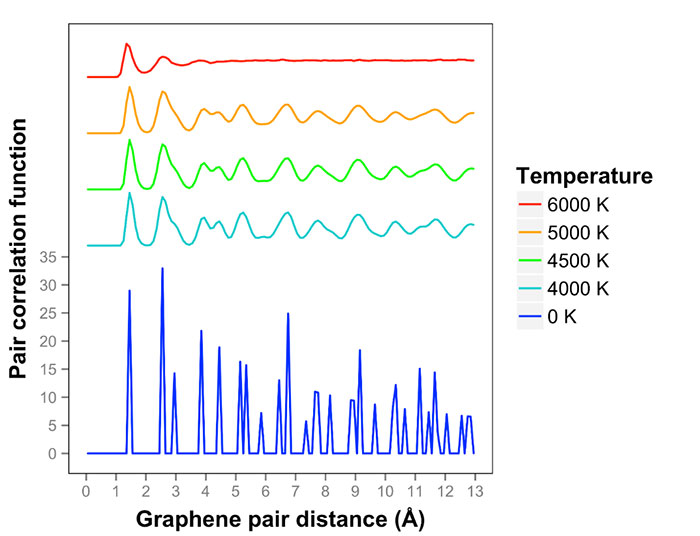
The team observed that after the 12 ps at 4000K, the surface had smooth oscillations and the lattice was maintained. Just a few 3-rings, due to neighboring atoms oscillating towards each other, were observed. No net diffusion was recorded at 4000 K. At 4500 K, the surface was displaying out of phase long-range oscillations, but the lattice was maintained over the 18ps simulation time. This may have been because the surface was not melted at that time or it was in a superheated condition. Longer simulation time might help answer the question. At 4500 K, the system appeared to be in a quasi-2D liquid state.
At 5000 K, the surface showed the initial stages of melting after 20 ps. The surface was observed to be rougher, but most atoms were still in 6-rings. Two diffusion events had occurred and one 5775 defect had nucleated. At 6000 K, the system had already melted after 7 ps, although it had not yet reached equilibrium. At this point, the system consisted 1D double-bonded chains. Pair correlation functions and Lindemann criteria were calculated.
The outcomes of this study sets the pace for longer and larger theoretical computations. These results may also inspire novel experimental studies of these high temperature materials along with other high temperature bulk alloys, or carbon nanotubes. These future experimental tests of melting could potentially be carried out in the weightless environment of space.



Reference
Eric Ganz, Ariel B. Ganz, Li-Ming Yang and Matthew Dornfeld. The initial stages of melting of graphene between 4000 K and 6000 K. Phys. Chem. Chem. Phys., 2017, 19, 3756—3762.
Go To Phys. Chem. Chem. Phys.