
Fischer–Tropsch synthesis has become such a familiar route for turning coal-, biomass-, or gas-derived syngas into fuels that it’s easy to forget how many practical hurdles remain beneath the surface. One issue keeps resurfacing no matter how the reaction is configured: carbon gradually builds up on the catalyst. Iron-based systems are especially prone to this, even though they excel under hydrogen-lean conditions and naturally steer the product slate toward olefins. At high temperatures, the very phases that make these catalysts so effective—most notably the iron carbides responsible for CO activation—also create conditions where amorphous and graphitic carbon accumulate far too quickly. When that buildup begins to outrun hydrogenation, active sites disappear and methane formation creeps upward. Researchers have tried to work around this through promoter tuning, tailored supports, or activation steps, but those solutions often end up complicating synthesis workflows or limiting scalability in industrial settings. To this end, new research paper published in Journal of Fuel Chemistry and Technology and conducted by Dr. Yan Chen, Dr. Huanhuan He, Dr. Yufeng Li, Dr. Bing Liu, and led by Professor Xiaohao Liu from the Department of Chemical Engineering, School of Chemical and Material Engineering at Jiangnan University, the researchers developed two physically mixed catalyst systems in which FeZnNa was combined either with oxide granules or with finely ground oxide powders. These configurations allowed them to isolate how interfacial contact governs CO dissociation, carburization rate, and carbon deposition. The powder-mixed SiO₂ and MgO systems emerged as highly stable catalysts because their intimate contact with Fe altered electron density and slowed excessive carbide formation.
The research team began with a FeZnNa catalyst synthesized through a co-precipitation route followed by Na impregnation, producing a ZnFe₂O₄-based precursor that could carburize into active iron carbides during FTS. To explore how oxide additives alter its behavior, they prepared two categories of mixtures. In the first, catalyst granules were blended with similarly sized oxide particles such as quartz sand or SiO₂ spheres. In the second, oxide powders were physically co-ground with the Fe catalyst before pelletization, forming more intimate contact interfaces. These materials were then activated under hydrogen and evaluated at 320 °C and 1 MPa syngas with H₂/CO = 1, allowing direct comparison of deactivation trajectories. The authors found that when quartz sand was used as a purely inert diluent, CO conversion collapsed from ~93% to ~15% within 20 h, accompanied by a steady rise in methane selectivity. This rapid decline reflected unchecked carburization: Mössbauer spectroscopy later confirmed that nearly the entire Fe phase transformed into χ-Fe₅C₂, with no residual Fe oxides remaining. In contrast, adding SiO₂ as granules slowed deactivation but did not eliminate it. The decisive change occurred when SiO₂ or MgO powders were co-ground with the catalyst. In these powder-mixed samples, CO conversion remained remarkably stable—95.6% for P-SiO₂ and 97.6% for P-MgO—without any perceptible downward drift over the same reaction period. They evaluated how oxides modulate Fe’s electronic environment and its susceptibility to carbon buildup and performed XRD and Mössbauer analyses which showed that P-SiO₂ and P-MgO contained a mixture of Fe₅C₂ and Fe₃O₄ after reaction, which indicated that carburization proceeded more slowly. TGA supported this conclusion: carbon deposition decreased in the sequence G-QS ≫ G-SiO₂ > P-MgO > P-SiO₂, with P-SiO₂ showing net mass gain dominated by oxidation of remaining Fe₃O₄ rather than carbon burn-off. Moreover, the team performed O₂-TPO and Raman spectra and both techniques revealed dramatically reduced low- and high-temperature carbon signals for P-SiO₂ and P-MgO, showing that these additives suppress both near-surface and polymerized carbon species. H₂-TPR added another layer of insight: mixing with SiO₂ or MgO shifted reduction peaks to higher temperatures, an indicator of stronger metal–oxide interactions that restrain Fe reducibility. CO-TPD further demonstrated that these oxides attenuate CO dissociation by reducing high-temperature desorption signals. Finally, XPS confirmed an electronic shift: Fe 2p₃/₂ peaks moved to higher binding energies in powder-mixed catalysts, revealing electron withdrawal by the oxide additives and a decreased tendency for Fe to drive aggressive CO dissociation.
In conclusion, Professor Xiaohao Liu and colleagues demonstrated that oxides can reshape the surface chemistry of Fe-based catalysts in ways that are both measurable and industrially meaningful. One important implication is how sensitive FTS performance is to the oxide–iron interface. Granular mixing, which limits points of contact, produces only modest gains in stability. Once oxide powders are physically integrated at a finer scale, the electronic and chemical environment at the Fe sites shifts enough to slow CO dissociation and mitigate excessive carburization. This observation underscores an often overlooked aspect of catalyst design: materials do not need covalent integration to influence one another, provided they can interact electronically or perturb surface adsorption equilibria. The identification of SiO₂ and MgO as particularly effective modifiers adds nuance to long-standing discussions about acid–base effects in FTS. Both oxides alter Fe’s electron density in a direction that weakens CO dissociation, yet they differ in morphology and chemical character. Their shared effectiveness suggests that it is the capacity to redistribute electron density—rather than acidity alone—that determines whether an oxide will slow carbon nucleation. This finding can be extended to other catalyst systems where excessive carburization, coking, or carbide formation undermines long-term activity. Several mechanistic threads converge in this work. XPS and CO-TPD imply that the oxide additives reduce Fe’s ability to populate antibonding orbitals in adsorbed CO, thereby lowering the rate of dissociative adsorption. Mössbauer spectroscopy shows that the resulting carburization pathway becomes more gradual, preventing the system from entering the rapid carbon-accumulation regime typical of χ-Fe₅C₂–dominated catalysts. TGA, Raman, and O₂-TPO all confirm that surface carbon is not only less abundant but structurally less evolved. The collective conclusion is that the powder-mixed oxides disrupt the feedback loop in which aggressive CO dissociation breeds rapid carbon deposition, which then accelerates further deactivation. For industrial FTS, where catalyst lifetimes directly affect economic viability, a low-complexity method that yields stable high conversions is noteworthy. The approach is readily scalable, avoids complex synthesis routes, and allows tailoring through oxide selection. Moreover, the conceptual framework emerging from this study—linking interfacial electron modulation with carburization control—may guide the development of next-generation Fe catalysts designed for carbon-lean operation at high temperatures. In a nutshell, the experimental results of Professor Xiaohao Liu and colleagues introduce a powerful strategy for tuning Fe-based catalyst stability without altering the intrinsic chemical composition of the catalyst.
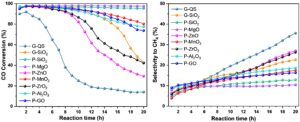
Fig. 1 Regulating catalytic performance of FeZnNa catalyst by mechanically mixing strategy with various oxides



Reference
Yan CHEN, Huanhuan HE, Yufeng LI, Bing LIU, Xiaohao LIU, Study on the regulation of Fischer-Tropsch synthesis catalytic performance by mixing oxides with iron-based catalysts, Journal of Fuel Chemistry and Technology, Volume 53, Issue 8, 2025, Pages 1212-1222,
