
SIGNIFICANCE
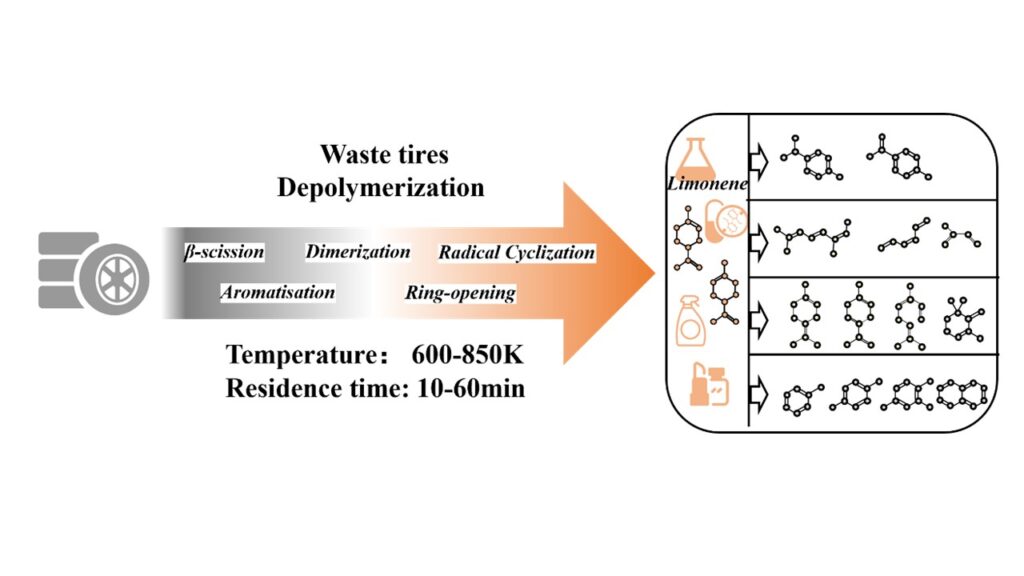
Around the world, mountains of worn-out tires stack up behind garages, clog municipal dumps, and quietly accumulate in industrial stockpiles—until the numbers reveal the scale of the issue. Global waste-tire volumes are projected to reach 27 million tons per year by 2030, prompting growing interest in treating discarded tires not as a liability but as a potential chemical resource. Pyrolysis stands out in this shift: instead of burning or grinding rubber into low-value fillers, it breaks polymers apart under oxygen-free heat, producing oils, gases, and carbon residues. Among these products, the unexpected presence of limonene, a high-value compound used in resins, solvents, and fragrances, has captured researchers’ attention. Yet the scientific picture behind limonene’s formation remains murky. Tires are chemically complex mixtures of natural rubber, SBR, BR, additives, sulfur networks, and carbon black, all of which crack, cyclize, and rearrange when heated. Past studies—often relying on static GC–MS snapshots—struggled to capture the fast-moving intermediates inside pyrolysis reactors, leading to inconsistent conclusions about when limonene peaks or why it later declines.
In recent research published in the peer-reviewed Journal Fuel, PhD candidate Yusong Zhang, Xingdong Li, Wensheng Xie, Yongming Lu,Xin Wang, Lei Zhang, Guozhao Ji, Yuan Gao, led by Professor Aimin Li from the Dalian University of Technology have explored a contribution in clarifying the mechanisms governing limonene formation and degradation during tire pyrolysis. They constructed a controlled tubular furnace system where nitrogen flow, sample mass, and heating rate were stabilized. They prepared waste tire granules to uniform particle size, which eliminated mass-transfer artifacts. The authors performed parallel pyrolysis runs to ensure reproducibility, while gas, liquid, and solid fractions were recovered through staged condensation, desiccation, filtration, and volumetric measurement. Multiple analyses probed evolving product chemistry in real time. The authors also performed thermogravimetry which showed decomposition onsets well below temperatures previously assumed, while TG–MS tracking of m/z=93 signaled the first emergence of limonene fragments near 550 K, fading at ~750 K. This directly contradicted older assumptions that limonene forms only in higher thermal windows. The team conducted STA-GC–MS and detected limonene across 550–750 K and identified 650 K as a production peak (a temperature at which FTIR spectra also showed strong unsaturated hydrocarbon formation, consistent with natural rubber degradation) and with temperatures approach 750 K, they found limonene yields dropped sharply; char decreased and gas fractions increased, indicating heightened bond scission and rearrangement. They also conducted GC–MS which demonstrated that limonene’s ratio decreased steadily with temperature, while aromatics and cycloolefins increased, implying transformation rather than volatilization. Statistical correlation analysis (SPSS) confirmed these relationships, revealing a near-perfect negative correlation between limonene and aromatic hydrocarbons (C7–C10). Moreover, the authors found that residence time testing further refined this dynamic. At 750 K, extending vapor residence from 10 minutes to 60 minutes halved pyrolysis oil yield while tripling gas output, showing intensified cracking. The limonene fraction decreased progressively, indicating that time amplified secondary reactions, including aromatization and Diels–Alder condensation leading to polycyclic aromatic hydrocarbons. Interestingly, longer residence suppressed intramolecular cyclization of alkatrienes, preventing accumulation of C10 cycloolefins and diverting pathways toward small olefins and PAHs. Moreover, Py-GC–MS added finer resolution by exposing the selective conversion of L-limonene to D-limonene below 700 K, followed by loss of L-limonene entirely above that threshold. Peak yields of L-limonene occurred at 650 K, whereas D-limonene peaked later, at 750 K. This contrast implied that modest heating favored stereoisomer stability, while stronger heating promoted rearrangement and cracking.
In conclusion, this work of Professor Aimin Li and colleagues developed new frameworks which showed that limonene is a transient intermediate rather than a stable pyrolysis product. Their work identifies how stereoisomer conversion and radical pathways govern yield decline, establishing design principles for reactors that intentionally preserve limonene. These findings reposition pyrolysis from indiscriminate thermal destruction toward engineered recovery of specialty chemicals. The team successfully identified limonene’s lifespan, its isomer interconversion, and the precise temperature at which disappearance accelerates by integrating in-line spectroscopic observation and their demonstration that D-limonene dominates because L-limonene undergoes early cracking provides a concrete rationale for stereoisomer behavior, which allow processor designs to intentionally target or suppress isomer pathways.


Reference
Yusong Zhang, Xingdong Li, Wensheng Xie, Yongming Lu, Xin Wang, Lei Zhang, Guozhao Ji, Yuan Gao, Aimin Li, Pyrolysis behavior and production characteristics of limonene in tire pyrolysis: Implications for waste valorization, Fuel, Volume 390, 2025, 134663,
