
Significance
The pursuit to increase the efficiency of low cost solar-to-electric energy conversion has led to the development of organic photovoltaics. Such advances are hoped to enable fabrication of larger and flexible devices in comparison with convectional inorganic photovoltaics. Such auspicious developments have however encountered multiple challenges in organic photovoltaics regarding their efficiency, scalability, cost and flexibility in order to effectively compete with inorganic and hybrid photovoltaics. Consequently, development of a large number of novel materials in the fields of organic photovoltaics and organic electronics has led to an optimization challenge for the selection process. Presently, fullerenes and fullerene derivatives, have shown to have very good mechanical characteristics while preserving the required electronic properties. Currently, comprehension of the transport and thermo-mechanical properties in pure phases has rapidly evolved, nonetheless, there is a need for evaluating at a fundamental level how having mixtures of molecules based on similar conjugated moieties, such as those of fullerenes, affects the macroscopic properties.
Recently, Dr. Naga Rajesh Tummala, Dr. Veaceslav Coropceanu and Professor Jean-Luc Bredas from the School of Chemistry and Biochemistry and Center for Organic Photonics and Electronics Georgia Institute of Technology and Professor Saadullah Aziz from King Abdulaziz University assessed and described how mixing affects the molecular packing, mechanical properties, and electronic parameters of interest for solar-cell applications. They also quantified the electronic and mechanical characteristics of mixtures of fullerenes by using a combination of molecular dynamics simulations and density functional theory calculations, following established procedures for pure phases of fullerenes. Their research work is published in Journal of Materials Chemistry C.
The research method employed the GROMACS software to perform molecular dynamics simulations of the various selected fullerenes. These simulations were based on the all-atom optimized potentials for liquid simulations force field to represent the Lennard-Jones and point-charge parameters for the carbon, oxygen, and hydrogen atoms. The morphologies derived from molecular dynamics simulations were then used as input for electronic-structure calculations performed at the density functional theory level with an optimally tuned range-separated hybrid functional.
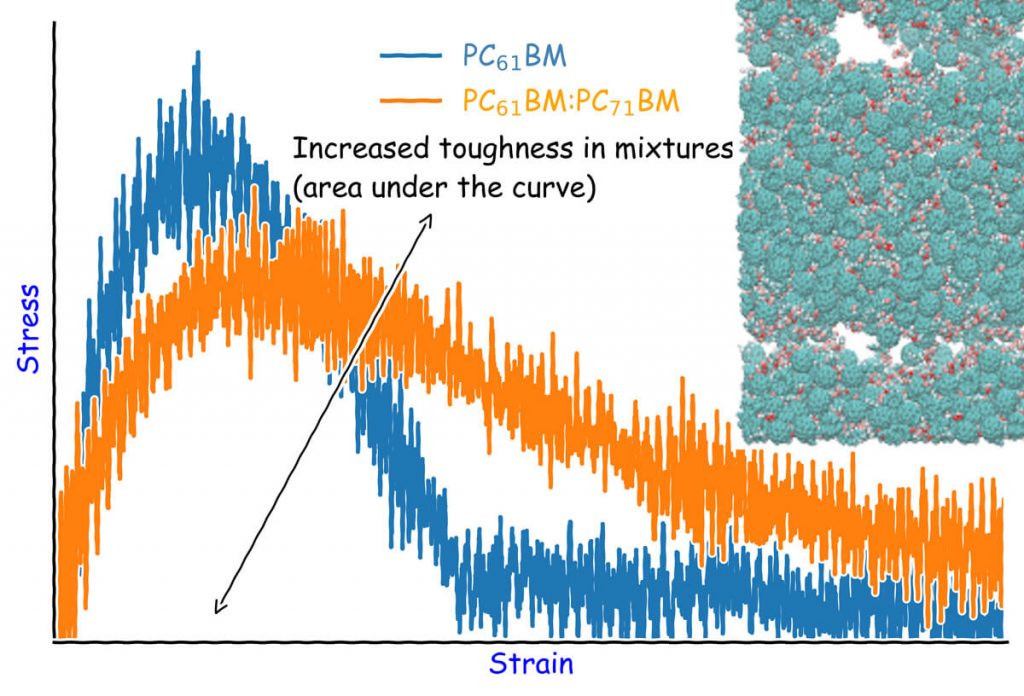
The authors observed that at less than 10% mole fraction, C70 fullerenes could not form a percolation network in the mixtures of C60:C70, however, the formation of small domains was observed. They also found out that the mixtures of fullerenes show increased fracture energy and similar tensile modulus and fracture toughness, indicating that the mechanical properties of the parent system were not compromised.
In summary, the study by Jean-Luc Bredas and colleagues presented an in-depth quantification of the packing, mechanical properties, and electronic disorder and partial density of states of mixtures of fullerenes and fullerene derivatives. Generally, they observed that the doping of PC61BM with PC71BM leads to the formation of shallower trap states than in the case of doping C60 with C70 and C84. Altogether, the results obtained by the Georgia Institute of Technology and King Abdulaziz University scientists can serve as a guide for future investigations of electronic disorder in various mixtures of small organic molecules exploited in organic electronics.




Reference
Naga Rajesh Tummala, Saadullah G. Aziz, Veaceslav Coropceanu, Jean-Luc Bredas. Characterization of the structural, mechanical, and electronic properties of fullerene mixtures a molecular simulations description. Journal of Materials Chemistry C, 2018, volume 6, page 3642.
Go To Journal of Materials Chemistry C