
Significance Statement
Raman spectroscopy as well as vibrational infrared spectroscopy is of ultimate importance in describing properties and structural geometries of nanostructures. Tackling multiradical and metallic systems in particular, using quantum chemical methods, proves to be very difficult. The problem originates from the fact that the single-reference wave functions lacks the capacity to suitably characterize such complicated electronic configurations. Although there are models that have been developed, and are powerful in this context, their application is computationally very demanding.
A conceptually more simple yet efficient solution for extended molecular systems with complicated electronic structure is associated with the use of fractional occupation numbers for the open-shell molecular orbitals. Although much efforts have been devoted towards the use of fractional occupation numbers in quantum chemical methods, geometrical second-order derivatives that include such fractional occupation numbers are yet to be developed. For this reason, theoretical spectroscopic analyses of open-shell molecular systems are very limited and restricted to small molecular systems.
In a recent research published in Chemical Physics Letters, Dr. Yoshio Nishimoto and Professor Stephan Irle both at Nagoya University in Japan derived, for the first time, a geometrical second-order derivative of the Mermin free energy function for the density-functional tight-binding (DFTB) approach. Their extension was applicable for all spin-restricted wave functions, which includes multi-radical, radical and metallic systems.
The authors implemented the fractional occupation number DFTB approach with energy and geometrical first and second order derivatives into a version of the GAMESS-US program package. They adopted an electron density convergence criterion of 10-9 for all numerical derivatives and 10-12 for all other computations. They optimized all structures with maximum and root mean square gradient thresholds of 10-9 and 1/3×10-9, respectively.
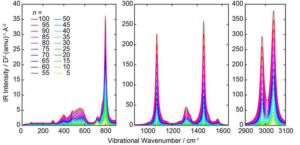
The authors employed and tested two existing sets of DFTB parameters; one was the “mio” set while the other was “slkoopt”. The convergence of the equations was achieved when the maximum difference of the Mulliken population derivatives between consecutive cycles was below 10-6. However, the authors observed that the tightening thresholds did not yield any change in the observed results. They computed the Raman activities by calculating the atomic forces and the second-order derivatives based on a finite electric field.
The applicability of the developed model was tested for nitronyl nitroxide radical systems. However, the orbital occupation of the singly occupied molecular orbital was exactly one, irrespective of the molecular structure.
Using traditional, first principles density functional theory (DFT) overestimated the vibrational frequencies. Therefore, the authors scaled DFT wavenumbers by 0.976. For the DFTB frequencies, the authors used scaling factors of 0.952 and 0.959 for ‘mio’ and ‘slkoopt’, respectively. This was in a bid to minimize root mean square deviations for the two sets.
The authors recorded a root mean square deviation of 67.56 cm-1 for the scaled DFT approach. The unscaled DFTB approach yielded root mean square deviations of 79.45 and 68.85 cm-1 for the ‘mio’ and ‘slkoopt’ parameters respectively, hence are comparable in accuracy with DFT but about three orders of magnitude faster. The newly introduced program is, therefore, compatible with computer systems with over 3000 basis functions, or roughly more than 1000 atoms. A relevant and preceding study about the computation of non-resonance Raman activity spectra is also published in The Journal of Chemical Physics.
References
Yoshio Nishimoto1,2 and Stephan Irle1,3. Quantum chemical prediction of vibrational spectra of large molecular systems with radical or metallic electronic structure. Chemical Physics Letters, volume 667 (2017), pages 317–321.
[expand title=”Show Affiliations”]- Department of Chemistry, Graduate School of Science, Nagoya University, Nagoya 464-8602, Japan
- Fukui Institute for Fundamental Chemistry, Kyoto University, 34-4 Takano Nishihiraki-cho, Sakyo-ku, Kyoto 606-8103, Japan
- Institute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Nagoya 464-8602, Japan
Go To Chemical Physics Letters
Yoshio Nishimoto. Analytic hyperpolarizability and polarizability derivative with fractional occupation numbers for large extended systems. The Journal of Chemical Physics, volume 146 (2017), page 084101.
Go To The Journal of Chemical Physics
