
Significance
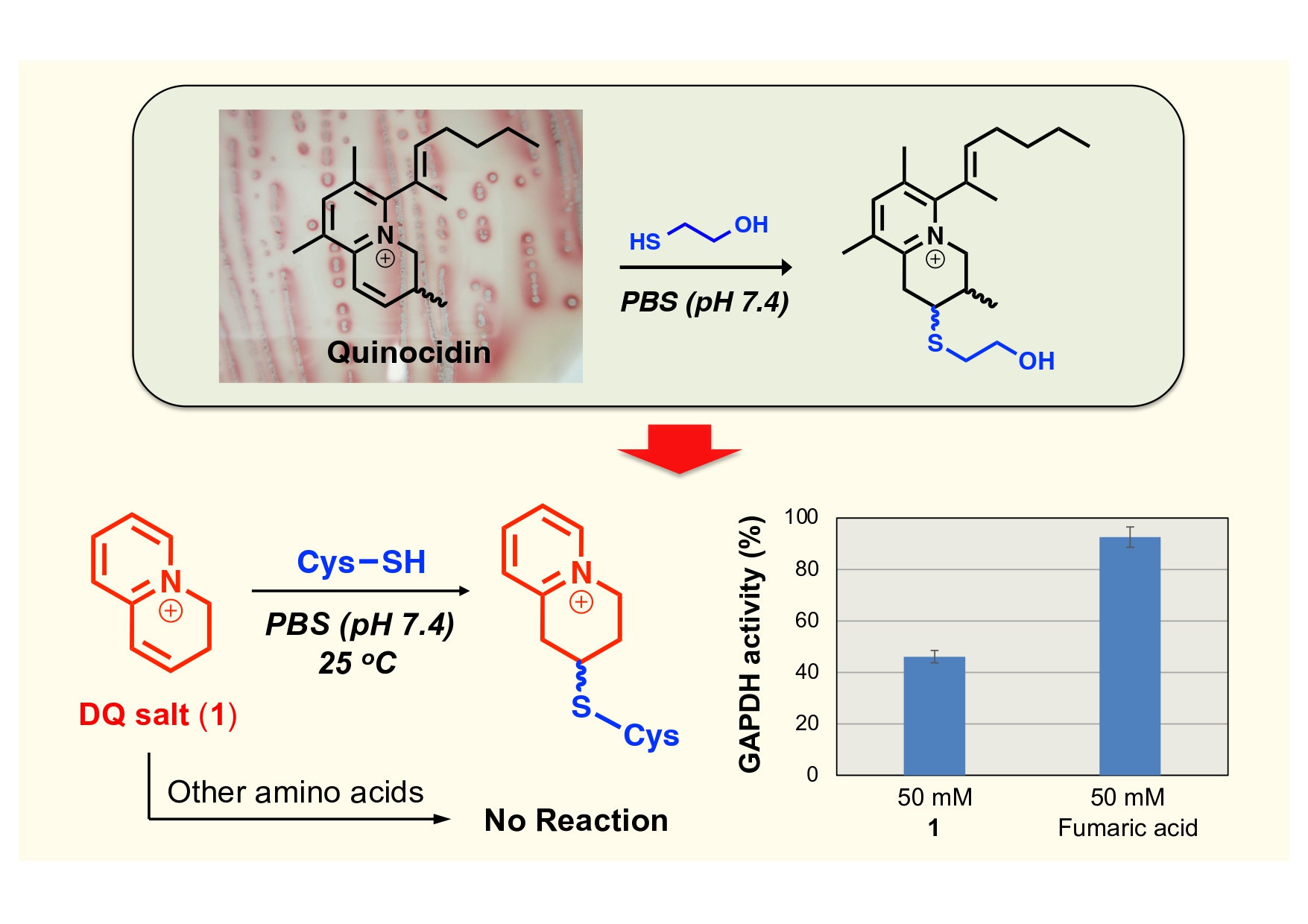
The team began by synthesizing the model compound DQ salt (1) in three concise steps from 3-butyn-1-ol. Sonogashira coupling with 2-bromopyridine produced an alkynyl intermediate, which was partially hydrogenated and cyclized through mesylation to close the 3,4-dihydroquinolizinium ring. The overall yield was moderate but reproducible, and the compound was purified by HPLC under trifluoroacetic acid conditions, yielding a stable, water-soluble salt suitable for biochemical assays. When the DQ salt was mixed with cysteine in phosphate-buffered saline (pH 7.4) at room temperature, the reaction proceeded almost instantaneously, with the adduct formation complete within ten minutes as confirmed by HPLC. NMR and high-resolution mass spectrometry validated a Michael-type addition mechanism at the β-position of the DQ ring. They found under identical conditions, the DQ salt was completely inert toward the other nineteen proteinogenic amino acids, including lysine, serine, and histidine, even after extended incubation. Afterward, the authors assessed the reactivity of the DQ ring toward more complex substrates using a series of capped cysteine-containing dipeptides. In each case, the electrophile reacted cleanly to generate the expected Michael adducts, although at slightly slower rates than with free cysteine. The reduced reaction rate was attributed to steric hindrance and to the higher pKa of the thiol group within the peptide context. Importantly, the reaction rates showed little dependence on the neighboring amino acid, suggesting that the DQ moiety can engage Cys residues independently of local sequence environment—a desirable feature for protein modification chemistry. The authors performed further validation came with glutathione (GSH), the canonical thiol model in biological systems and found the DQ salt readily formed an adduct with GSH but showed no reaction with the oxidized disulfide form (GSSG), confirming its exclusive reactivity toward free thiol groups. Encouraged by these results, the researchers extended the test to the 17-mer peptide odorranalectin, which contains two cysteine residues. Upon incubation, nearly all peptide was converted into either a DQ–peptide adduct or an oxidized disulfide form, confirming that the DQ electrophile could access cysteine residues within larger, folded peptides. Finally, to assess biochemical relevance, the DQ salt was incubated with GAPDH, an enzyme possessing an essential cysteine in its active site. At higher concentrations (50 mM), compound 1 significantly inhibited enzyme activity, achieving over 50% inhibition within three hours—substantially greater than that achieved by fumaric acid, a known thiol-reactive Michael acceptor.
In conclusion, Professor Dr. Yu Nakagawa and colleagues new models establish the DQ ring as a new class of mild, cysteine-selective electrophiles suitable for biochemical and pharmaceutical exploration. They provided convincing evidence that the 3,4-dihydroquinolizinium ring is an autonomous, tunable electrophile capable of engaging thiols under benign aqueous conditions, by isolating the minimal reactive unit of the natural product quinocidin. The implications extend beyond mere reagent development. The DQ scaffold introduces a permanent positive charge at the modification site, a property that could alter the electronic landscape of proteins in a predictable way. Such charge-modulating modifications could enable the design of novel biochemical tools for probing redox regulation or stabilizing specific protein conformations. Moreover, since the DQ ring can be readily synthesized and potentially derivatized, it provides a foundation for building small-molecule covalent inhibitors or fluorogenic probes targeting catalytic cysteines. In the broader context of medicinal chemistry, cysteine-targeted covalent drugs have re-emerged as a major frontier, from kinase inhibitors to antiviral agents. However, the success of such drugs depends critically on controlling reactivity to minimize off-target modification. The DQ ring, with its moderate but precise electrophilicity, could fill this gap between sluggish and overly aggressive electrophiles. The fact that it inhibited GAPDH, an archetypal thiol enzyme, hints at the potential to modulate other redox-active proteins involved in metabolism or oxidative stress responses. The study also highlights the power of natural product chemistry in inspiring new chemical logic. Quinocidin’s unusual ring system, once a structural curiosity, now stands as a template for rational electrophile design. Their findings validate the DQ core as a reactive center and open a conceptual path toward designing positively charged Michael acceptors with tunable reactivity. In a nutshell, the 3,4-dihydroquinolizinium ring emerges from the new study as a compact, selective, and synthetically accessible platform for cysteine modification and covalent enzyme inhibition. Indeed, the new discovery expands the chemist’s repertoire of thiol-reactive motifs and may, in time, underpin a new generation of site-specific protein conjugation or redox-modulating drugs.

REFERENCE
Nakagawa Y, Manabe Y, Kondo W, Kondo T, Irie K. 3,4-Dihydroquinolizinium Ring, the Core Structure of Quinocidin, as a Cysteine-Selective Electrophile. Chempluschem. 2025;90(7):e202500149. doi: 10.1002/cplu.202500149.
Chempluschem.