Significance Statement
Ionic liquids are becoming popular owing to a wide range of characteristics and amazing groundbreaking developments in biology, materials science, chemistry, and physics. The possibility of adequately designing these systems at the molecular level has attracted numerous research work aiming at understanding them from a perspective of optimizing and manipulating their chemical design in a bid to obtain large scale properties. Molecular simulation allows for a molecular based understanding of ionic liquid large scale properties, therefore, presenting a powerful tool for development and use of ionic liquids.
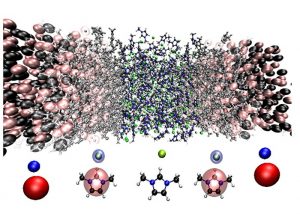
Advanced resolution method allows for obtaining full atomistic details of a selected region and coupling it with a region with generic molecular model, coarse-grained, without atomistic details, that functions as a reservoir of energy as well as particles. This indicates that grand canonical-like advanced resolution can be implemented as a tool for identifying the essential atomistic degree of freedom needed to get a certain property.
The study aimed at evaluating the feasibility of implementing the grand canonical-like advanced resolution to ionic liquids. However, the challenging part would be the presence of explicit ions and the ability to build a coarse-grained procedure for a physically valid system-reservoir coupling. Therefore, Dr. Christian Krekeler and Professor Luigi Delle Site at Freie Universität of Berlin proposed two different methods for developing an accurate coarse-grained model. One method focused on a coarse-grained model which did not carry charges but reproduced the ion-ion radial distribution functions of a full atomistic simulation. In the other approach, the ions in the coarse grained model carried also charges. Their work is now published in Physical Chemistry Chemical Physics.
Simulations performed in the study was using the package GROMACS. Researchers adopted 1,3-dimethylimidazolium chloride as the test system on the basis that it’s complex enough to test the robustness of the proposed simulation method and conversely simpler, therefore, allowing for a large number of numerical analyses in a simple way without expensive computational efforts.
The researchers set up two systems. The first one with 350 ion pairs was applied to derive two coarse grained potentials which were then transferred to a larger system with over 1000 ion pairs. Simulation temperature was set at 400K and a 2fs time step. The electrostatic interactions were computed through particle mesh Ewald method. They prove that the grand-canonical simulation can be applied to ionic liquids, despite the challenging conceptual and computational aspects of the system.
The grand canonical-like advanced resolution allows for the treatment of atomistic spherical regions embedded in a large reservoir of structureless molecules, therefore, the dependence of dynamic and structural properties as a function of size of the atomistic region can be systematically analyzed. By analyzing the results obtained from various simulations defined by different size atomistic regions, it will be possible to establish the connection between time and length scales of given properties. For instance, one could calculate the hydrogen bond-hydrogen bond autocorrelation function expressed as a function of size of the atomistic region.
Comparison between the results obtained with various ionic liquids would be necessary in clarifying the effect of specific molecular chemical structure of these ions. This study paves way for a direct check and an in-depth understanding of the hypothesis of rattling of ions in long-living ion cages.


Reference
Christian Krekeler and Luigi Delle Site. Towards open boundary molecular dynamics simulation of ionic liquids. Phys. Chem. Chem. Phys., 2017, 19, 4701—4709.
Go To Phys. Chem. Chem. Phys.