Significance
Polymer networks stiffen, soften, densify, or fail according to how their internal domains rearrange under stress and time, yet most synthetic systems cannot keep changing that internal organization once fabrication ends. Living matter does something quite different. Its condensed phases appear, reorganize, merge, and sometimes disappear as molecular interactions shift during growth or response, and those rearrangements directly alter function. In a recent research paper published in Advanced Materials, Dr. Cheng Liu, Dr. Chaowei He, Dr. Xiaobin Dai, Prof. Li-Tang Yan, and Prof. Huaping Xu from Tsinghua University, developed a hydrogel system in which visible-light-activated diselenide units initiate in situ polymerization of methacrylic acid from an existing PMAAm-based network, generating PMAAc dangling chains that drive a timed sequence of phase generation, separation, and fusion. They also developed a spatially addressable route for building multi-region hydrogels with different local moduli using visible-light patterning.
The scientific problem is not simply how to make a hydrogel stronger. Soft materials already offer many routes for raising modulus or toughness through extra crosslinks, chain entanglement, filler addition, or dehydration. The harder question is how to build a material whose internal compatibility keeps changing after the material has been formed, so that one mechanical state does not merely switch to another, but evolves through a sequence of structurally distinct states. That issue has remained difficult to address because ordinary polymer phases arise from fixed segmental incompatibility or fixed attractive interactions. Once the network is made, the system usually settles into a stable distribution of domains or requires continuous energy input to hold a temporary one. Static chemistry produces static phase logic.
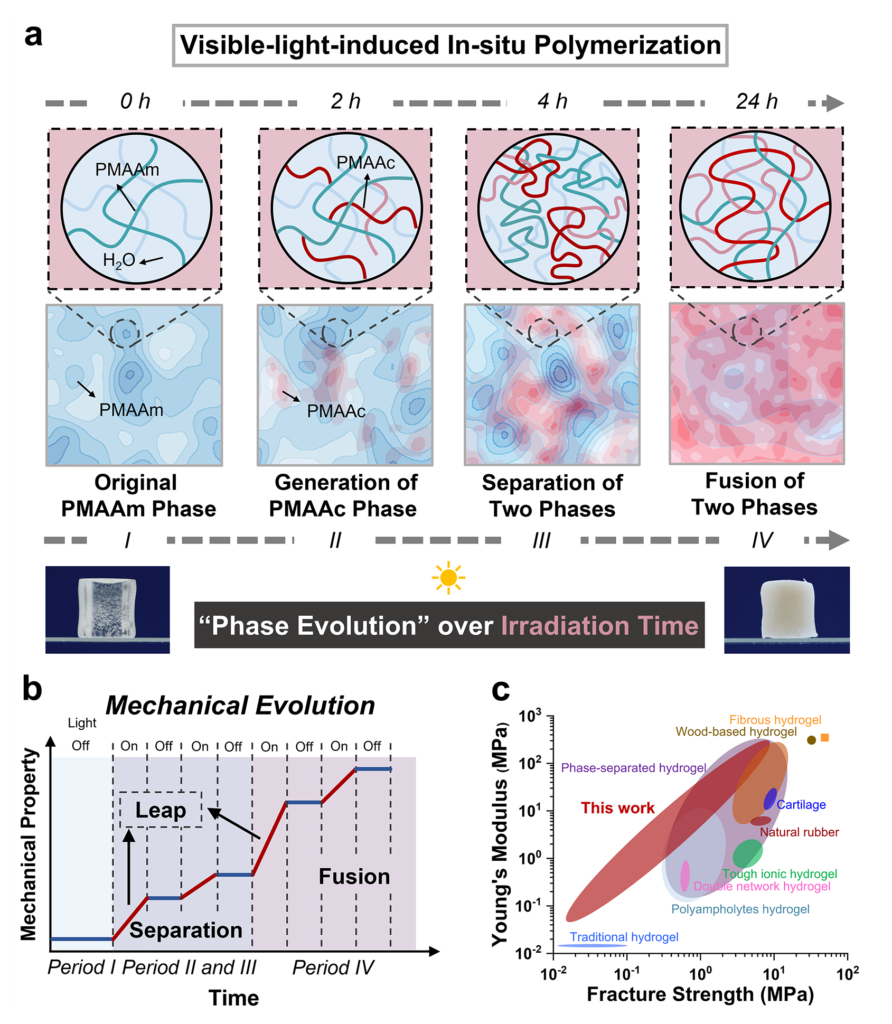
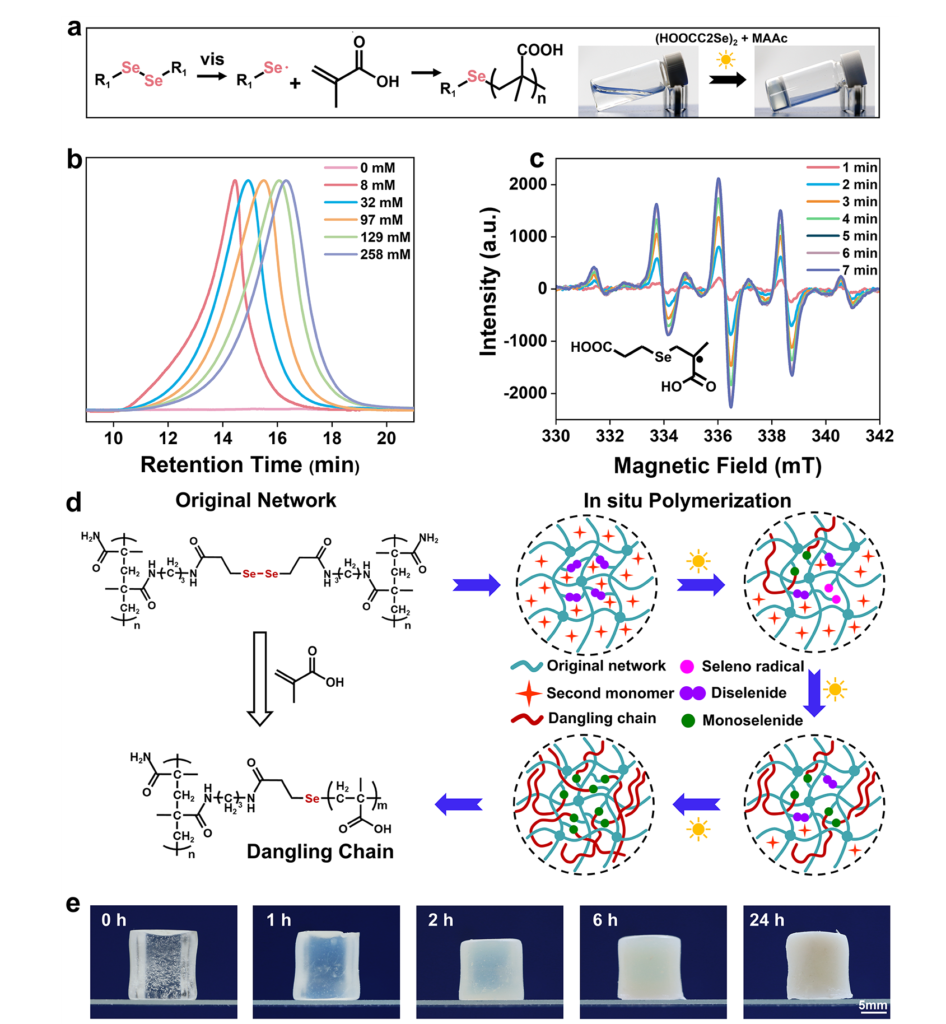
The hydrogel platform chosen for the study makes that reasoning especially compelling. Hydrogels already contain large amounts of water, mobile segments, and mechanically important mesoscale heterogeneity, so phase behavior can reshape load transfer in a way that is easy to feel and measure. In their design, methacrylic acid monomer resides within a preformed polymethyl acrylamide-based network, while diselenide units embedded in the network generate radicals under visible light and start in situ polymerization. The key expectation is not merely that new polymethacrylic acid chains will form, but that these dangling chains will first develop their own association tendencies and later interact more strongly with the host network through evolving hydrogen-bond patterns. That makes the central challenge a question of competing kinetics and thermodynamics: local phase formation can occur quickly as new chains appear, whereas deeper chain rearrangement and interphase merging demand more time because mobility and entanglement impose a real penalty on structural reorganization.
The research team first established that the diselenide chemistry could genuinely initiate methacrylic acid polymerization under visible light. NMR, GPC, and EPR measurements all supported the formation of PMAAc and the presence of propagating radical species, while the radical signal responded directly to illumination and decayed more slowly in the dark. That controllable initiation step matters for more than chemical proof. If initiation were too fast, the material would jump through structural states before they could be separated experimentally or regulated spatially; if too slow, phase reorganization would lose practical control. The reported initiation rate supplied a usable window in which the internal state of the gel could be steered by irradiation time.
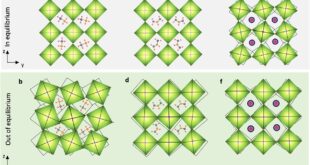
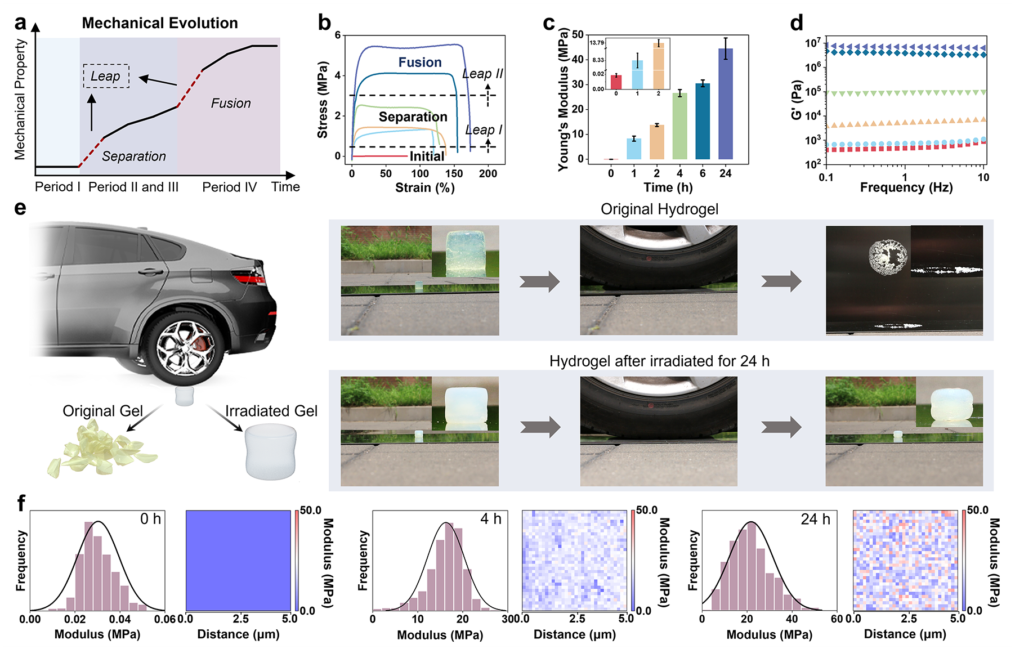
Once the authors built the hydrogel containing the PMAAm-based network, MAAc, and diselenide crosslinking points, they observed an immediate macroscopic clue that the internal structure was changing: a transparent gel gradually turned opaque during irradiation. The investigators interpreted that loss of transparency as refractive-index heterogeneity created by newly formed domains, which is a sensible reading because the chemical network remained singular while the internal composition grew less uniform. The new PMAAc existed as dangling chains covalently attached to the original network, so the system did not become a simple blend of two disconnected polymers. That detail is important. Because the new chains stayed tethered, the material could separate internally without falling apart, and later fusion of phases could still feed into a continuous load-bearing framework.
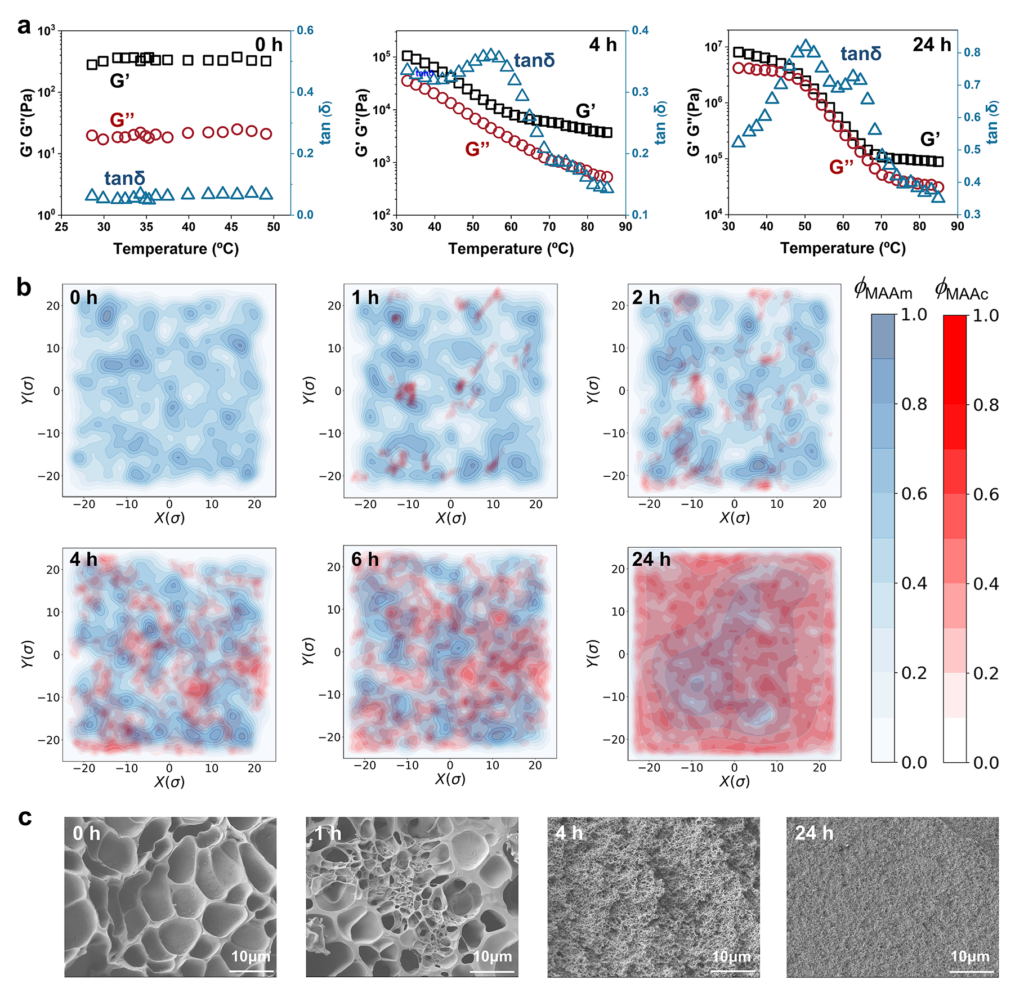
The study examined the temporal sequence of those structural changes with several complementary methods. Rheology revealed that a new phase with its own glass transition emerged after irradiation, and prolonged exposure later produced two glass-transition features consistent with a fused but compositionally uneven state. Low-field NMR tracked the increasing proportion of a mixed PMAAc-PMAAm phase in the later period, while SAXS registered rising heterogeneity during the early hours and declining heterogeneity at longer times. It generated a new PMAAc-rich phase, sharpened separation from the original PMAAm-rich environment, and then moved toward a fused state in which both polymers remained enriched in different local ratios. That later merger is especially revealing because it shows that temporal order in soft matter can emerge from a moving balance between newly created incompatibility and stronger final association.
The researchers reinforced that interpretation with coarse-grained and all-atom molecular dynamics simulations, which placed the strongest phase separation near the early stage and the fusion-dominant regime later, around 6 to 24 hours. SEM images echoed the same story in structural terms: the original freeze-dried gel looked porous and loosely packed, intermediate states displayed denser regions coexisting with looser ones, and the long-irradiated material became denser and more uniform. A denser but more even internal structure changes mechanics in a very concrete way. Stress is less likely to concentrate at sharp density boundaries when interfaces are reduced, so the material can transmit load across the body more effectively. The paper never treats morphology as decoration; morphology is the route by which chemistry becomes mechanics.
To explain why the phases appeared in that order, the authors focused on hydrogen-bond evolution among carboxylic acid and amide groups. Chemical disruption of those interactions softened and clarified the gel, linking the immiscible domains to hydrogen-bonded association. FTIR and WAXS then showed that the bonding population did not stay fixed during irradiation. Small amounts of newly formed PMAAc initially associated with PMAAm, increasing PMAAc content later favored COOH-COOH interactions and PMAAc-rich domains, and continued evolution drove the system toward the more stable COOH-CONH2 associations that promoted fusion of PMAAc-rich and PMAAm-rich regions. Here the trade-off becomes very clear. Short-time behavior follows the pace of polymer growth, which favors local PMAAc-domain formation before large-scale rearrangement can catch up. Long-time behavior follows the search for the most stable hydrogen-bond network, which pulls the system toward interphase mixing and fusion. The sequence is ordered because chain motion is slow enough to let kinetic and thermodynamic preferences act in different windows.
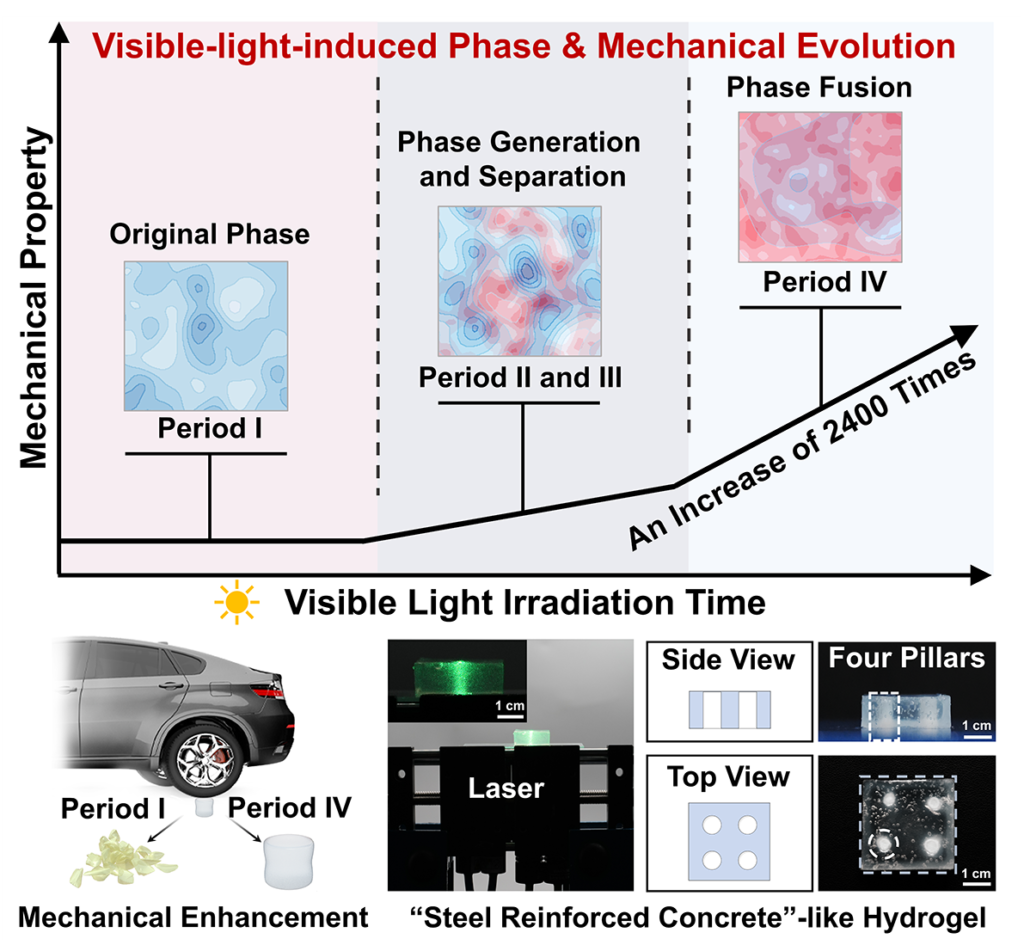
Tensile modulus rose from 18.5 kPa to 44.5 MPa, compressive modulus climbed from 57.7 kPa to 22.2 MPa, and the increases in storage modulus and fracture energy were similarly dramatic. The authors also identified two pronounced jumps in stiffness during irradiation, one associated with the onset of phase generation or separation and another with the onset of phase fusion. A single monotonic stiffening curve could be explained by growing polymer content, but step-like gains aligned with phase transitions argue that internal structural reorganization, not just added solid fraction, governs the change in load-bearing behavior. The material stiffens in qualitatively different ways at different times: early domain formation introduces new physical crosslinking and dissipation pathways, while later fusion produces a denser and more uniform route for stress distribution.
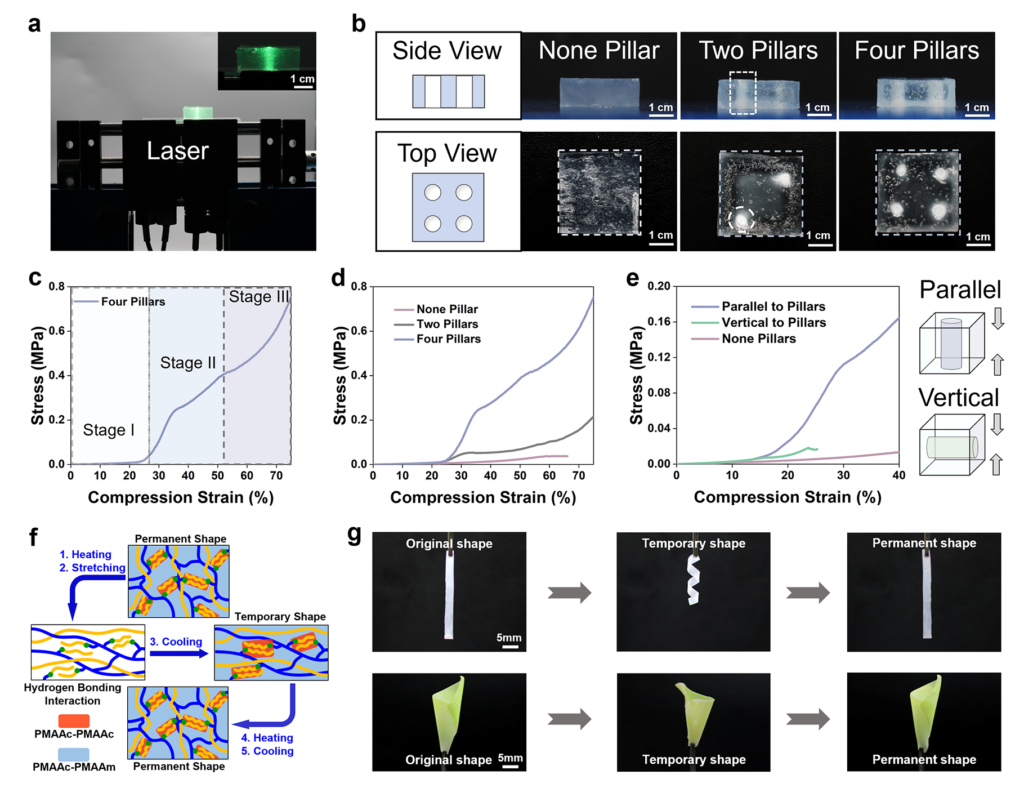
The work of Professor Huaping Xu and colleagues also widened the argument beyond bulk strengthening. Visible light penetrated the hydrogel far more uniformly than UV-based initiation, which allowed the authors to generate fairly even modulus increases through thickness and across surface regions. By using patterned irradiation, they created a “steel reinforced concrete”-like hydrogel with stiff pillars inside a softer matrix, and the resulting composite displayed staged stress response and directional mechanical behavior. The same underlying chemistry also gave the irradiated gel shape-memory capability through hydrogen-bond exchange and reformation. These demonstrations matter because they convert the study from a story about a single tough gel into a broader design principle: if phase evolution can be positioned in space as well as in time, then modulus is no longer a fixed material property but a programmable field inside one connected body which mean that internal phase history can be treated as a controllable design parameter for soft composites, adaptive structures, and locally reinforced architectures.

Reference
Liu, Cheng & He, Chaowei & Dai, Xiaobin & Yan, Li‐Tang & Xu, Huaping. (2025). Achieving Mechanical Evolution in Polymer Materials Through Phase Evolution Induced by Visible Light. Advanced Materials. 37. 10.1002/adma.202508549.