Journal of Alloys and Compounds, 27 March 2013.
Takeshi Kobayashi, Akito Takasaki
Graduate School of Engineering and Science, Shibaura Institute of Technology, 3-7-5 Toyosu, koto-ku, 135-8548 Tokyo, Japan
Department of Engineering Science and Mechanics, Shibaura Institute of Technology, 3-7-5 Toyosu, koto-ku, 135-8548 Tokyo, Japan
Abstract
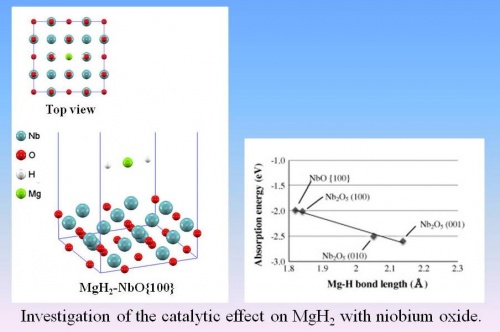
In this paper, in order to investigate the dependence of oxidation state of metal oxides, adsorption reaction of a MgH2 on Nb2O5 ((1 0 0), (0 1 0), (0 0 1)) and NbO {1 0 0} surfaces, in which the valence states of the Nb are +5 and +2, respectively, were mainly investigated using periodic Density Functional Theory (DFT) calculation. Mg–H bond length, adsorption energy and Local Density of States (Local DOS) were calculated.
As a result, the Mg–H bond length in adsorption process on niobium oxides enlarged as compared with optimized MgH2, because H atom came closer to Nb atom and created hybrid orbital. Moreover, Mg–H bond length of MgH2 cluster with Nb2O5 became longer than that with NbO. The longer chemical bonds generally imply weaker bond strength. It is suggested that this might be affected by not only valence state of Nb but also a lattice configuration of catalysts. However, adsorption energy of MgH2 with Nb2O5 was lower than that of NbO. It is considered that the interaction of MgH2 with Nb2O5 was stronger than that with NbO like Local DOS results. Our results indicate that the lower oxidation state of Nb is catalytically active on the adsorption process of MgH2.
Additional information
We carried out theoretical calculations based on Density Functional Theory (DFT) method to investigate the dependence of oxidation state of niobium oxide in magnesium hydride.