Organometallics, 2015, 34 (1), pp 242–253.
Gagik G. Melikyan *, Rhoda Hughes , Bianca Rivas , Kellyanne Duncan , Nare Sahakyan.
Department of Chemistry and Biochemistry, California State University Northridge, Northridge, California 91330 United States.
Abstract
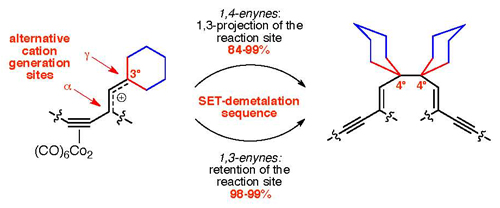
Radical coupling reactions of Co2(CO)6-complexed 1,4-enynes occur in a regio- and stereoselective fashion, providing access to 3E,7E-decadiene-1,9-diynes in excellent yields (84–99%). The formation of contiguous quaternary carbon atoms follows a tandem allylic rearrangement that projects an original reaction site gamma to the metal core. Propargyl alcohols with an α-alkenyl group as a substituent are treated with HBF4, followed by the reduction of the highly conjugated propargyl cations with zinc. The scope of the reaction is expanded to include 1,4-enyne complexes with cyclic and acyclic substituents gamma to the metal core, as well as aliphatic and aromatic substituents attached to the acetylenic termini. The alternative design includes relocation of the cation generation site—α-to-γ—prior to the reduction step, employing either the cation isolation technique with HBF4 or an in situ generation of ionic propargyl triflates with Tf2O. Retention of the reaction site in 1,3-enynes is observed in both γ-alcohols and γ-Me ethers, affording respective γ,γ-radical dimers in excellent yields (98–99%).
Copyright © 2015 American Chemical Society.
Related article: Melikyan G. G. Propargyl Radical Chemistry: Renaissance Instigated by Metal Coordination. Acc. Chem. Res. 2015, 48, 1065-1079.
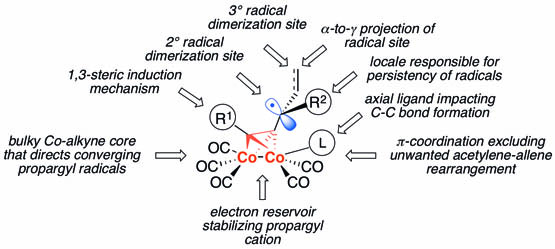
Over the last two decades, radical chemistry of propargyl systems was developed into a potent synthetic field providing access to classes of organic compounds that are otherwise hardly accessible. The levels of diastereoselection thus achieved (up to 100%) are unprecedented for free propargyl radicals as well as for organic radicals pi-bonded to transition metals. These advances were enabled by the coordination of the triple bond to a Co2(CO)6-core that precluded an acetylene-allene rearrangement, stabilized requisite propargyl cations, created conformational constraints at the carbon-carbon bond formation site, configurationally altered the acetylenic moiety allowing for 1,3-steric induction, increased bulkiness of propargyl triads thus controlling the spatial orientation of converging radicals, allowed for —α-to-γ— projection of the reaction site and alteration of the transiency of radical intermediates. In the course of these studies, a number of popular “beliefs” were proven to be untrue. First, cobalt-complexed propargyl cations – which have long been considered to be thermally labile species – were engaged in synthetically meaningful transformation at temperatures as high as 147°C. Second, in radical dimerization reactions, higher reaction temperatures didn’t adversely impact the yields and levels of d,l-diastereoselectivity. Third, pi-bonded organometallic radicals were effectively controlled with complementary mechanistic tools, thus achieving the highest levels of stereoselectivity (up to 100%) in inter- and intramolecular reactions. Fourth, meso stereoisomers, being thermally labile and kinetically disfavored, were discovered to be major products in intramolecular cyclizations induced by cobaltocene. A concept of “sequestered” propargyl radicals was introduced to explain disparity in diastereoselectivity data: heterogeneous reducing agents allegedly produce “free” radicals, while homogeneous reductants generate “sequestered” radicals associated with reductant-derived oxidized species. In intramolecular reactions, a d,l-to-meso reversal of stereoselectivity was discovered with zinc being replaced with cobaltocene as a reductant. Two novel reactions that belong to a new field of “unorthodox organometallic radical chemistry” were discovered: the spontaneous conversion of cobalt-complexed propargyl cations to radicals and the THF-mediated process wherein a THF molecule assumes a new role of an initiator in radical reactions. Novel mechanistic tools and methodologies for controlling stereoselectivity in radical reactions can now be expanded toward new types of pi-bonded unsaturated units (dienes, arenes, diynes, enynes) and transition metals other than cobalt (Fe, Cr, Mo, W, Mn). Novel stereoselective methods provide access to topologically and functionally diverse 3,4-diaryl/dialkyl-1,5-alkadiynes, 3,4-disubstituted 1,5-cycloalkadiynes (C8-C12), 3,4-dialkoxy-1,5-(cyclo)alkadiynes, and 3,7-diene-1,9-alkadiynes which can be used in targeted syntheses of organic assemblies of relevance to medicinal chemistry, materials science, and natural product syntheses.